Mise au point
Publié le 14 déc 2016Lecture 9 min
Tachycardies ventriculaires catécholergiques
A. LEENHARDT, I. DENJOY, E. VARLET, A. MESSALI, P. DEJODE, F. EXTRAMIANA, Département de rythmologie, Hôpital Bichat, Paris
Les tachycardies ventriculaires catécholergiques (TVC) sont des arythmies d’origine héréditaire, engageant le pronostic vital, caractérisées par l’apparition de troubles du rythme ventriculaire polymorphes dans le contexte d’une stimulation adrénergique (exercice physique, émotion forte). Elles font partie des maladies rares avec une prévalence mal connue, estimée à 1/10 000. Même s’il s’agit d’une entité rare, le diagnostic est d’une importance vitale car ces arythmies sont responsables d’un taux important de mort subite chez le sujet jeune. Les autopsies moléculaires de cas de morts subites non expliquées ont retrouvé des mutations pathogènes de RYR2 dans 15 % des cas chez des sujets de moins de 40 ans. Chez des patients non traités, la mortalité peut atteindre un taux de 30 % avant l’âge de 40 ans.
Des progrès importants ont été réalisés ces dernières années justifiant cette mise au point sur l’état actuel des connaissances.
Caractéristiques cliniques
Les porteurs d’une TVC ont un coeur structurellement normal et un ECG 12 dérivations normal, hormis un certain degré de bradycardie sinusale chez 20 % des patients. Il est possible d’observer chez certains une fibrillation atriale qui peut précéder la survenue des arythmies ventriculaires. La plupart des patients présentent des symptômes dans l’enfance ou dans l’adolescence, mais des cas plus atypiques existent dans la 3e ou 4e décennie.
Le diagnostic
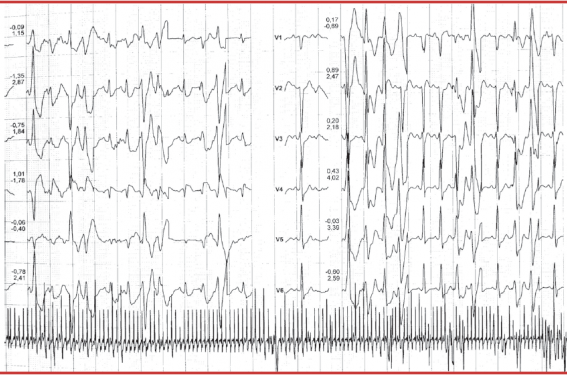
Il se fait lors d’un stress intense ou à l’effort où l’on voit apparaître, sur l’ECG, des extrasystoles ventriculaires (ESV) dans un premier temps, isolées et souvent monomorphes. Ces ESV apparaissent souvent ensuite bigéminées puis polymorphes avec des formes répétitives, doublets, triplets puis la survenue de salves à mesure que l’effort se poursuit. Ces arythmies ventriculaires disparaissent lorsque l’exercice ou l’effort est stoppé. Le seuil de déclenchement des arythmies ventriculaires est assez souvent reproductible, se situant aux alentours de 110 à 130/min. La morphologie des ESV est le plus souvent à type de retard gauche, axe inférieur mais parfois à type de retard droit, axe supérieur (figure). L’apparition de tachycardies ventriculaires bidirectionnelles est très caractéristique du diagnostic de TVC mais est assez peu fréquente, avec une rotation de l’axe du QRS à 180° un battement sur deux. Les autres conditions dans lesquelles ces tachycardies ventriculaires bidirectionnelles peuvent apparaître sont l’intoxication digitalique et le syndrome d’Andersen-Tawil.
Figure. Survenue au cours d’une épreuve d’effort d’ESV polymorphes et d’une salve de TV polymorphes chez un patient porteur d’une TVC.
Le diagnostic de TVC est posé en présence d’un cœur structurellement normal, d’un ECG normal et d’une arythmie ventriculaire survenant à l’exercice ou sous stimulation catécholergique, sous forme de tachycardie ventriculaire bidirectionnelle ou d’ESV polymorphes ou de tachycardies ventriculaires polymorphes chez un sujet de moins de 40 ans ou chez un sujet porteur d’une mutation pathogène dans un des gènes connus pour donner des TVC. Le diagnostic peut être également fait chez des membres de la famille d’un sujet atteint qui présentent des troubles du rythme ventriculaires induits par l’exercice ou des tachycardies ventriculaires en l’absence de cardiopathie structurelle. Chez les sujets de plus de 40 ans, on retient ce diagnostic lorsque l’on a pu éliminer une coronaropathie.
Le test d’effort est donc l’outil diagnostique le plus utile pour cette pathologie rythmique. Chez les sujets asymptomatiques la spécificité est de 97 %, mais la sensibilité est faible, voisine de 50 % pour prédire la présence de mutations dans la famille. Lorsque le test d’effort n’est pas possible, un Holter peut être utile. Les tests pharmacologiques à l’isoprénaline ou à l’adrénaline sont également utiles. Ces tests restent globalement peu sensibles mais très spécifiques.
Les tests génétiques sont largement recommandés pour confirmer le diagnostic et si la recherche de mutation est positive, elle sera à réaliser chez tous les membres de la famille au premier degré. Dans le cas où la recherche génétique serait négative chez un patient porteur d’une TVC, les apparentés au 1er degré doivent bénéficier d’épreuves d’effort répétées, selon leur âge, à la recherche d’arythmie ventriculaire d’effort.
Aspects électrophysiologique et génétique
Le caractère familial a été décrit pour la première fois en 1960 par Berg et la première grande série a été publiée en 1995 par notre équipe.
C’est en 1997 qu’une équipe finlandaise a mis en évidence le caractère autosomique dominant de cette pathologie liée à une mutation sur le chromosome 1. Cette mutation a été découverte en 2001 sur un gène codant pour le récepteur cardiaque de la ryanodine cardiaque (RYR2). Les mutations dans RYR2 couvrent à peu près 65 % des cas de TVC. RYR2 est impliqué dans le métabolisme du calcium intracellulaire et joue un rôle important dans le couplage excitation-contraction. Les mutations dans RYR2 entraînent un relargage diastolique du calcium intracellulaire à partir du réticulum sarcoplasmique. Cette augmentation du calcium cytosolique active l’échangeur sodium-calcium entraînant un courant entrant de calcium qui provoque des postdépolarisations tardives, ce qui entraîne des arythmies ventriculaires déclenchées. Ces afflux de calcium intracellulaire diastolique sont encore plus prononcés en cas d’hypertonie adrénergique.
Toujours en 2001, une mutation autosomique récessive a été découverte dans un gène codant pour la calsequestrine cardiaque (CASQ2). Ces mutations représentent 2 à 5 % des cas de TVC. La calsequestrine cardiaque est une protéine qui agit sur le stock calcique intracardiaque avec un effet inhibiteur sur RYR2.
D’autres mutations ont été depuis décrites, beaucoup plus rares, dans le gène codant pour la triadine, la calmoduline et encore plus rarement l’ankyrine B et KCNJ2. Dans ces deux derniers cas, les mutations sont en général associées avec un syndrome du QT long congénital et il pourrait donc s’agir de phénocopies des TVC.
Au total, à peu près un tiers des patients porteurs de TVC n’ont pour l’instant pas de mutation génétique retrouvée.
La recherche de mutation pour RYR2 et CASQ2 est recommandée chez tous les patients chez lesquels il existe une suspicion clinique importante de TVC. Lorsqu’une mutation est authentifiée, la recherche de cette mutation chez les membres apparentés au 1er degré de la famille est recommandée. Il a été mis en évidence récemment le fait qu’il y avait de nombreux variants de RYR2, non pathogènes, qui sont authentifiés chez ces sujets. Ceci est très important à prendre en compte, afin de ne pas porter à tort un diagnostic génétique erroné de TVC.
Stratification du risque
La stratification du risque pose un problème difficile dans les TVC. Actuellement, on considère comme des marqueurs de risque le fait d’avoir des signes cliniques à un très jeune âge, une histoire d’arrêt cardiaque récupérée et l’absence de traitement bêtabloqueur.
Il n’y a pas actuellement de corrélation phénotype/génotype évidente comme elle existe dans le syndrome du QT long. Néanmoins, il semble qu’une mutation dans la partie C terminale de RYR2 soit associée à davantage de troubles du rythme ventriculaire non soutenus qu’une mutation dans la partie N terminale. Enfin, les patients porteurs d’une mutation de CASQ2 semblent avoir un phénotype un peu plus sévère que les patients porteurs d’une mutation de RYR2.
Traitement
Une recommandation valable pour tous les patients porteurs de TVC est d’éviter les exercices physiques intenses et le sport de compétition. L’autre point très important est la nécessité absolue d’une observance thérapeutique parfaite. La mauvaise observance est dans notre expérience responsable d’un nombre très significatif d’événements cardiaques graves.
Le traitement bêtabloquant
Le traitement le plus important, de première ligne, reste le traitement bêtabloquant à la dose la plus importante tolérée. Les recommandations actuelles sont de traiter tous les patients symptomatiques et les porteurs d’une mutation pathogène même s’ils sont asymptomatiques. Le traitement doit être surveillé en évaluant les patients par une épreuve d’effort ou un Holter à l’effort. Il n’y a pas d’étude permettant de comparer de manière scientifiquement indiscutable les différents bêtabloquants, néanmoins le nadolol paraît supérieur aux autres dans différentes études non randomisées, à une posologie supérieure à 1,5 mg/kg/j. Une explication possible de l’effet favorable de cette molécule serait son effet chronotrope plus prononcé que les autres bêtabloquants, avec une demi-vie plus longue.
L’association à la flécaïnide acétate
Quand le traitement bêtabloquant est insuffisant pour prévenir la survenue d’arythmie ventriculaire à l’exercice, l’association à la flécaïnide acétate (Flécaïne, Meda) a été proposée (recommandation de niveau IIa). Des études encore limitées en termes de nombres de patients ont montré des résultats tout à fait intéressants de cette association. Il n’y a encore que très peu d’expérience dans l’utilisation de la flécaïnide acétate en monothérapie chez des patients qui sont, soit intolérants, soit qui ont des effets secondaires importants sous bêtabloquants. L’utilisation en routine de la flécaïnide acétate en monothérapie n’est actuellement pas recommandée en l’absence de preuve clinique de son efficacité. La flécaïnide acétate agit probablement directement au niveau d’un canal sodique (NAV1.5) mais a également un effet direct bloquant sur le canal RYR2.
La dénervation sympathique
La dénervation sympathique gauche est une technique chirurgicale dans laquelle les deux tiers inférieurs du ganglion stellaire gauche ainsi que les ganglions thoraciques T2 à T4 sont ablatés. Ce sont des sources importantes de norépinéphrine 5 pour le cœur. La complication la plus fréquente est le syndrome de Claude Bernard Horner dont la fréquence a nettement diminué avec l’utilisation de la vidéothoracoscopie et qui, dans l’ensemble, est très souvent réversible. L’indication est actuellement réservée à des patients qui restent symptomatiques malgré un traitement médical maximum, associant bêtabloquant et flécaïnide ou qui sont intolérants aux bêtabloquants ou chez lesquels les bêtabloquants sont contre-indiqués.
Le défibrillateur automatique (DAI)
Il est actuellement recommandé chez les patients qui ont un arrêt cardiaque sous traitement ou des syncopes récidivantes, ou des troubles du rythme ventriculaire récidivants, malgré une thérapeutique médicale optimale. Dans notre expérience personnelle, nous n’avons aucun décès de patient appareillé avec un DAI. Néanmoins, il y a de très nombreuses complications liées en particulier à des chocs inappropriés survenus sur des arythmies atriales ou des problèmes techniques comme des ruptures de sonde ou des surdétections. Deux études ont par ailleurs mis en évidence le fait que les chocs électriques pouvaient être inefficaces sur des arythmies ventriculaires assez spécifiques des TVC comme les TV bidirectionnelles.
Le traitement bêtabloquant et parfois la Flécaïne sont de toute façon toujours associés au DAI dont on comprend bien qu’il s’agit d’une indication thérapeutique qui ne doit être posée qu’avec une extrême précaution chez des sujets jeunes qui vont être exposés à des complications potentiellement importantes liées à l’appareil et aux sondes.
Parmi les perspectives thérapeutiques, outre certaines molécules qui pourraient un jour être proposées, agissant spécifiquement sur RYR2, des essais sont actuellement menés chez l’animal avec des thérapeutiques purement géniques qui seront peut-être un jour disponibles.
Conclusion
Des progrès importants ont été faits ces dernières années concernant le diagnostic et le traitement des patients porteurs de TVC. Le point important est surtout de faire le diagnostic devant des syncopes et des convulsions chez des sujets jeunes dont les caractéristiques sont de survenir à l’effort ou lors d’une émotion intense.
Le traitement repose toujours en premier lieu sur le traitement bêtabloquant, sur une bonne observance, sur une hygiène de vie avec l’abstention de tout effort violent et de toute compétition sportive.
Des progrès importants doivent être encore faits dans le domaine du diagnostic génétique, dans le domaine de la stratification du risque de cette maladie qui reste encore actuellement la plus grave des canalopathies.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :