

Publié le 16 oct 2023Lecture 16 min
Cardiomyopathies : les recommandations de l’ESC 2023
Raphaël COHEN, Unité INSERM 970, interne de cardiologie, Paris

Les recommandations ESC 2023 sur les cardiomyopathies (CMP) ont été présentées fin août à Amsterdam et publiées dans l’European Heart Journal dans le même temps. En dehors de la cardiomyopathie hypertrophique (recommandations ESC 2014), ces recommandations sont les premières étudiant les cardiomyopathies dans leur ensemble (de cause génétique ou non).
Plusieurs points essentiels ont été évoqués :
• Nosologie : plus que le génotype, le phénotype global (clinique, morphologie/fonction ventriculaire et génotype) permet une classification plus pertinente.
• Diagnostic : en plus de la clinique, de l’ECG et de l’échocardiographie, l’imagerie en coupe (IRM cardiaque et/ou scanner cardiaque) fait une entrée fracassante, permettant l’évaluation des infiltrations cicatricielles ventriculaires (fibrotiques et/ou adipeuses).
L’accent est mis sur le parcours de soins dès le plus jeune âge selon la présence ou non de symptôme ou d’un cas dans la famille en utilisant une approche multiparamétrique après une identification du phénotype pour arriver à un diagnostic étiologique. À noter une nouvelle entité : la cardiomyopathie non dilatée du ventricule gauche (CNDVG).
• Génétique : ces recommandations insistent sur la nécessité de se référer rapidement à un centre expert afin d’avoir une prise en charge génétique spécialisée permettant un diagnostic et une prise en charge familiale.
La prise en charge des symptômes, des troubles du rythme, et les préventions primaires et secondaires de la mort subite y sont détaillées.
• Risque de mort subite : ces recommandations viennent compléter celles de 2023 sur la mort subite, insistant sur la stratification du risque nécessaire pour l’indication à la pose d’un défibrillateur, avec des stratégies différentes selon le phénotype de la CMP.
Enfin, chaque cardiomyopathie est détaillée depuis son diagnostic jusqu’à sa prise en charge en insistant sur la CMH et la CMD : Dans la CMH, à noter l’entrée du mavacamten premier inhibiteur sélectif de la myosine cardiaque tandis que la place du tafamidis, un stabilisateur spécifique de la transthyrétine, est confirmée dans le traitement des formes symptomatiques de la cardiomyopathie amyloïde liée à la transthyrétine.
Il est impossible de rentrer dans les détails d’un document de plus de 145 pages. Certaines parties ne seront donc pas traitées ou brièvement résumées pour ses éléments essentiels : prise en charge pédiatrique, détails précis de la prise en charge génétique, cardiomyopathie arythmogène du ventricule droit (CAVD), certaines cardiomyopathies rares d’origine métaboliques (RASopathies, myopathies spécifiques). Pour cela, nous vous renvoyons au document original.
Définitions (tableau 1)
Cardiomyopathie : anomalie du myocarde structurellement et fonctionnellement anormal, en l’absence de coronaropathie, d’HTA, de valvulopathie et de cardiopathie congénitale suffisantes pour être à l’origine de l’anomalie myocardique observée.
Ne sont plus considérés comme des cardiomyopathies à proprement parler : la non-compaction du ventricule gauche (l’hypertrabéculation étant plutôt un trait phénotypique s’intégrant dans les différentes cardiomyopathies) et le syndrome de Tako-Tsubo.
Focus
Le phénotype CNDVG comprendra des individus qui, jusqu’à présent, ont été diversement décrits comme atteints de CMD (mais sans dilatation du ventricule gauche), de cardiomyopathie ventriculaire gauche arythmogène (CVGA), de CAVD à prédominance gauche ou de CMD arythmogène (mais souvent sans remplir les critères diagnostiques de la CAVD).
Les patients atteints de CMP doivent être pris en charge dans leur globalité avec différents acteurs de santé afin d’assurer un parcours de soins adéquat : cardiologues spécialisés, spécialistes en imagerie cardiaque, infirmiers, psychologues cliniciens, généticiens et autres spécialités médicales en fonction des atteintes extra-cardiaques (internistes, gastro-entérologues, etc.).
Prise en charge diagnostique
Ces recommandations insistent sur l’approche multiparamétrique à partir de la clinique et/ou de l’histoire familiale faisant suspecter une CMP qui sera confirmée par l’imagerie multimodale (figure 1) la classant dans un ou des phénotypes suscités.
Clinique et prélèvements biologiques
La présentation clinique initiale est variée :
– insuffisance cardiaque, arythmies, syncope, douleur thoracique mettant à contribution les soins primaires (médecins généralistes, pédiatres) ;
– et/ou une histoire familiale ;
– découverte fortuite.
Une prise en charge systématique est donc nécessaire.
Certains signes cliniques (drapeaux rouges) doivent faire évoquer une CMP génétique et/ou syndromique.
(Tableau 2)
Les anomalies ECGs (bloc auriculo-ventriculaire, aspect de préexcitation ventriculaire, anomalies de repolarisation et des voltages de QRS élevés ou bas) des différents phénotypes et étiologies ne sont pas détaillées ici (tableau 7 des recommandations).
(Tableau 3)
Pour les examens de seconde intention (IIa C), se référer au tableau 8 du document princeps.
Imagerie
L’échocardiographie afin d’évaluer les dimensions des cavités cardiaques ainsi que les fonctions ventriculaires gauches/droites systoliques et diastoliques VG est recommandée pour tous les patients au diagnostic et durant le suivi pour monitorer la progression et aider à la prise en charge thérapeutique et la stratification du risque (I B).
(Tableau 4)
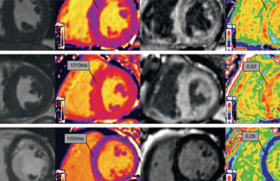
L’imagerie en coupe, en particulier l’IRM cardiaque, est désormais indispensable au diagnostic ainsi qu’au suivi des CMP.
En plus de communiquer des informations sur la morphologie et la fonction cardiaque, elle permet d’évaluer le rehaussement tardif correspondant à la fibrose myocardique ainsi que sa topographie. Le rehaussement tardif est un marqueur de progression de la CMP et un facteur majeur de risque de mort subite (MS).
L’IRM oriente vers des étiologies plus précises avec un apport bien supérieur à l’échocardiographie : CNDVG, CAVD, myocardite, amylose cardiaque, sarcoïdose, hémochromatose (surcharge en fer) (figure 2).
Un scanner cardiaque injecté doit être envisagé chez les patients suspects de CMP chez qui l’IRM est contre-indiquée (IIa) et/ou une cardiopathie congénitale ou une coronaropathie sont suspectées (IIa).
Par ailleurs :
– l’imagerie nucléaire utilisant un traceur osseux (biphosphonate – pyrophosphate) est recommandée chez tous les patients chez qui l’on suspecte une amylose ATTR avec atteinte cardiaque (I) ;
– un PET-TDM au FDG doit être envisagé pour le diagnostic chez les patients suspects d’une sarcoïdose cardiaque (IIa).
Enfin, la biopsie endomyocardique peut être envisagée pour aider au diagnostic et à la prise en charge lorsque les résultats d’autres examens cliniques suggèrent une inflammation, une infiltration ou une maladie de surcharge qui ne peut être identifiée par d’autres moyens (IIa).
(Figures 1 et 2)
Tests génétiques
Ces recommandations insistent très fortement sur la prise en charge génétique ayant un bénéfice direct pour confirmer le diagnostic du cas index, mais aussi des apparentés, éclairer le pronostic en particulier la stratification du risque rythmique, poser l’indication d’un traitement spécifique, et aider à la procréation.
L’architecture des CMP est complexe, on distingue (de manière très simplifiée) (figure 3) :
– atteinte monogénique autosomique dominante avec une pénétrance incomplète et une expression clinique variable ;
– atteinte oligo- ou polygénique de gravité variable selon le type et le nombre de variants mutés.
Principes généraux de prise en charge des patients atteints de cardiomyopathie
Insuffisance cardiaque
Les recommandations pour l’insuffisance cardiaque des CMP suivent les recommandations de l’ESC 2021, mises à jour en 2023, sans réel traitement spécifique au phénotype (figure 4).
Fibrillation atriale
La fibrillation atriale (FA) est l’arythmie la plus fréquente de tous les phénotypes de CMP et est associée à un risque accru d’événements cardio-emboliques, d’insuffisance cardiaque et de décès.
Traitement anticoagulant
L’anticoagulation peut être considérée chez les patients ayant une CMR (IIa) et chez les patients ayant une CMD, une CNDVG ou une CAVD avec un score CHA2DS2-VASc = 1 chez les hommes ou = 2 chez les femmes (IIa).
Contrôle du rythme vs contrôle de la fréquence
Comme dans les recommandations ESC sur la FA, l’ablation par cathéter est en 1re ligne. De même, l’ablation du nœud atrio-ventriculaire doit être envisagée si la stratégie de contrôle de la fréquence a été choisie, mais que la fréquence ventriculaire reste élevée avec ou sans symptômes.
Arythmies ventriculaires et risque de mort subite
Ces recommandations complètent celles de 2022 sur les arythmies ventriculaires et le risque de mort subite. Celles-ci préconisaient :
– en cas de TV incessantes ou d’orage rythmique : une ablation par cathéter de l’arythmie, si cela a été possible ou bien une modulation du système nerveux autonome (bloc sympathique) voir une assistance mécanique ventriculaire gauche ;
– en cas d’arythmies ventriculaires sur cicatrice, en plus de l’arsenal médicamenteux limité (bêta-bloquants, sotalol et amiodarone), d’avoir recours à des techniques d’ablation ou de neuromodulation spécifiques nécessitant une prise en charge dans un centre expert.
Défibrillateur implantable (DAI) et risque de mort subite (tableau 5)
L’évaluation du risque de mort subite est depuis quelques années bien plus précise, ceci grâce à de nouveaux scores de risque, dépendant du phénotype, mais aussi de la génétique (Applications ESC Pocket guidelines). Ils permettent de calculer le risque de mort subite à 5 ans, avec des seuils indiquant ou non l’implantation d’un DAI.
L’implantation d’un DAI est seulement recommandée si l’espérance de vie est > 1 an avec une bonne qualité de vie (I). La pose du DAI n’est pas une décision anodine (complications locales, infectieuses, chocs inappropriés, qualité de vie, conduite). Sa pose doit donc être guidée par une information éclairée du patient en prenant en considération ses convictions (I).
Si l’indication est posée, il faut évaluer le bénéfice du DAI (qualité de vie, bénéfice/risque) (I).
En l’absence d’indication à un pacing pour une bradycardie, un risque faible de la nécessité de pacing anti-tachycardique (ATP), et pas d’indication à une resynchronisation, un DAI sous cutané doit être envisagé à la place d’un DAI transveineux (IIa).
Suivi des patients
Les patients ayant une CMP doivent être suivis au long cours afin de détecter une évolution des symptômes, de la fonction ventriculaire et d’arythmies ainsi que le risque d’événements indésirables des traitements. La fréquence du suivi est déterminée par la gravité de la maladie, l’âge et les symptômes.
Examen clinique, avec un ECG 12 dérivations et une échocardiographie : tous les 1 à 2 ans, ou plus tôt si les patients se plaignent de nouveaux symptômes (I).
Un Holter ECG est recommandé tous les 1 à 2 ans chez la plupart des patients pour détecter les arythmies atriales et ventriculaires asymptomatiques, et en cas de syncope ou de palpitations (I).
Une IRM cardiaque doit être envisagée tous les 2 à 5 ans ou plus fréquemment chez les patients dont la maladie évolue (I).
L’épreuve d’effort cardio-pulmonaire (ECP avec mesure de la VO2 max peut fournir des preuves objectives de l’aggravation fonctionnelle de la maladie, mais ne doit être effectuée que tous les 2 à 3 ans, à moins qu’il n’y ait une évolution dans les symptômes. L’épreuve d’effort sur tapis roulant peut également fournir des informations fonctionnelles précieuses chez les patients incapables d’effectuer une ECP.
Exercice physique
Ces recommandations insistent sur la nécessité de maintenir une activité physique. La contre-indication quasi absolue n’est plus à l’ordre du jour. Un exercice physique même intense peut même être indiqué dans certaines CMP.
(Figure 6)
Phénotypes spécifiques de CMP
Cardiomyopathie hypertrophique
Plusieurs avancées notables dans la CMH.
L’évaluation du risque de mort subite a évolué pour intégrer deux facteurs de risque supplémentaires :
– un rehaussement tardif de plus de 15 % de la masse ventriculaire gauche ;
– FEVG < 50 %.
Chez les patients à faible risque, le DAI peut être discuté en présence de l’un de ces deux facteurs (classe IIb).
La préconisation d’une activité physique selon le génotype et le phénotype (ci-dessus).
Un nouveau traitement spécifique : le mavacamten, inhibiteur de la myosine cardiaque, est indiqué en 2e intention après utilisation des bêtabloquants ou du vérapamil (IIa) pour soulager les symptômes liés à une obstruction intraventriculaire gauche.
Diagnostic, évaluation et prise en charge symptomatique
Points clés
Diagnostic positif : épaisseur de la paroi du VG ≥ 15 mm sur n’importe quel segment du myocarde qui ne s’explique pas uniquement par les conditions de charge. Les degrés inférieurs d’épaississement de la paroi (13-14 mm) nécessitent l’évaluation des antécédents familiaux, des résultats génétiques et des anomalies de l’ECG.
Génétique : 50 % des CMH sont de transmission mendélienne dans la majorité autosomique dominante avec un risque de transmission de 50 %.
Focus
Obstruction intraventriculaire gauche :
Définition : gradient en Doppler continu maximal dans la voie d’éjection du ventricule gauche ≥ 30 mmHg, généralement un seuil ≥ 50 mmHg est retenu pour le traitement invasif (figure 7).
Mesure hygiénodiététique :
– éviter la déshydratation et la consommation excessive d’alcool ;
– la perte de poids doit être encouragée.
Thérapeutiques médicamenteuses (figure 8) :
– il n’existe pas d’ECT montrant un bénéfice sur la mortalité, il s’agit de recommandations empiriques afin d’améliorer les symptômes : le disopyramide, titré jusqu’à la dose maximale tolérée (habituellement 400-600 mg/jour), peut être ajouté aux bêtabloquants non vasodilatateurs (baisse du gradient – faible risque rythmique) ;
– si les bêtabloquants sont contre-indiqués : vérapamil (dose initiale de 40 mg trois fois par jour jusqu’à un maximum de 480 mg par jour). Attention si obstruction sévère > 100 mmHg et/ou HTAP (risque d’OAP).
Nouveauté : le mavacamten (inhibiteur cardiaque de la myosine ATPase).
NB :
Les dilatateurs artériels et veineux, y compris les nitrates et les inhibiteurs de la phosphodiestérase de type 5, peuvent exacerber l’obstruction intra-VG et doivent être évités si possible.
Les diurétiques de l’anse ou thiazidiques à faible dose peuvent être utilisés avec prudence pour améliorer la dyspnée associée à l’obstruction intra-VG, mais il est important de prévenir l’hypovolémie.
Les bêtabloquants seraient délétères si pas d’élévation gradient VG au repos ou provoqué. S’ils doivent être utilisés avec précautions si angor, préférer le vérapamil.
L’utilisation prudente de nitrates oraux peut être envisagée afin de diminuer les pressions de remplissage.
(Figures 7 et 8)
Traitement chirurgical : techniques de réduction septale (TRS)
Un traitement de réduction septale peut également être envisagé chez les patients présentant des symptômes légers (classe II de la NYHA) réfractaires au traitement médical, qui ont un gradient intra-VG au repos ou un gradient maximal provoqué ≥ 50 mmHg (à l’effort ou lors du Valsalva) et une régurgitation mitrale systolique moyenne à sévère liée au SAM, une FA ou une dilatation moyenne à sévère de l’oreillette gauche, dans des centres experts présentant des taux de complications procédurales faibles.
L’intervention chirurgicale la plus couramment pratiquée pour traiter l’obstruction dans la CMH est la myectomie du septum ventriculaire. Cette intervention permet d’abolir ou de réduire considérablement le gradient intra-VG dans plus de 90 % des cas, de réduire la régurgitation mitrale systolique liée au SAM et d’améliorer la capacité d’exercice ainsi que les symptômes.
Les déterminants préopératoires d’un bon résultat à long terme sont l’âge < 50 ans, la taille de l’oreillette gauche < 46 mm, l’absence de FA et le sexe masculin.
Prise en charge de l’insuffisance cardiaque et/ou de la FA en cas de CMH (figure 9)
Prévention de la mort subite dans la CMH (figure 10)
L’événement arythmique léthal le plus fréquemment enregistré est la fibrillation ventriculaire spontanée (FV), mais l’asystolie, le bloc AV et l’activité électrique sans pouls sont également décrits.
Les principaux déterminants de morts subites à évaluer afin de poser l’indication d’un DAI en prévention secondaire sont :
– antécédents : âge, TVNS, ATCD de mort subite dans la famille chez un patient jeune, syncope ;
– imagerie :
• échocardiographie : épaisseur maximale d’une paroi ventriculaire > 30 mm, gradient intra-VG élevé,
• IRM cardiaque : rehaussement tardif ≥ 15 %, anévrysme apical ;
– variants sarcomériques.
La Task Force maintient le principe de l’estimation du risque à l’aide de l’outil validé HCM Risk-SCD comme 1re étape de la prévention de la mort subite chez les patients âgés de 16 ans ou plus, et recommande l’utilisation d’un score de risque validé (HCM Risk-Kids) pour les enfants et les adolescents âgés de moins de 16 ans.
Cela contraste avec les recommandations 2020 de l’AHA/ACC pour le diagnostic et le traitement des patients atteints de CMH, dans lesquelles l’outil est considéré comme une aide au processus de prise de décision partagée pour la pose d’un DAI chez les patients présentant des marqueurs de risque cliniques.
(Figure 10)
Cardiomyopathie dilatée
La cardiomyopathie dilatée (CMD) est définie par la présence d’une dilatation du VG ET d’une dysfonction systolique inexpliquées uniquement par des conditions de charge anormales ou une maladie coronaire.
Diamètre télédiastolique ventriculaire gauche > 58 mm chez les hommes et > 52 mm chez les femmes ; et un volume télédiastolique indexé ≥ 75 mL/m2 chez les hommes et ≥ 62 mL/m2 chez les femmes en échocardiographie.
La dysfonction systolique globale du ventricule gauche est définie par une FEVG < 50 %.
Chez les apparentés d’un cas index :
La présence d’une dilatation isolée du ventricule gauche avec une fonction systolique préservée ou en présence d’un variant familial causal est suffisante pour diagnostiquer une CMD chez un parent.
Génétique :
Dans les cohortes contemporaines, des variants génétiques causaux sont présents chez 40 % des patients atteints de CMD, et entre 10 et 15 % dans les cas de CMD induite par la chimiothérapie, l’alcoolisme ou le péripartum.
La découverte d’un variant génétique causal chez un patient atteint de CMD permet de mieux prédire l’évolution de la maladie, de contribuer aux indications d’implantation de DAI, d’éclairer le conseil génétique et de permettre un dépistage familial pour les proches.
Certains variants sont à risque accru de mort subite et une IRM cardiaque doit être envisagée chez les parents même si la fonction ventriculaire gauche est normale.
(Tableaux 6 et 7)
Prévention de la mort subite dans la CMD (figure 11)
Facteurs de risques supplémentaires : syncope, rehaussement tardif à l’IRM.
Cardiopathie non dilatée du ventricule gauche
Génétique
Les gènes les plus couramment impliqués dans la CNDVG sont DSP (fibrose et poussées inflammatoires), FLNC, DES (desmosomes), LMNA ou PLN, mais il existe un chevauchement important avec le fond génétique de la CMD et de la CAVD
Caractéristiques ECG
Maladies neuromusculaires – sarcoïdose : PR prolongé ou bloc AV
Laminopathies : PR prolongé, FA, ESV et présentent fréquemment un microvoltage dans les dérivations précordiales
Prévention de la mort subite (figure 11)
Idem CMD avec niveau de preuve moindre.
Cardiomyopathie arythmogène du ventricule droit
Elle se caractérise structurellement par une atrophie myocardique progressive avec remplacement fibro-graisseux du myocarde du ventricule droit. Des lésions peuvent également être présentes dans le myocarde ventriculaire gauche ; une maladie prédominante du ventricule gauche peut coexister dans la même famille. Le diagnostic se fait selon les critères publiés en 2010 par Marcus et coll.
Prise en charge antiarythmique
Prévention de la mort subite (figure 12)
Cardiomyopathie restrictive (figure 13)
Étiologies et diagnostic
La CMR est caractérisée par une physiologie restrictive due à une atteinte myocardique et/ou endocardique avec le plus souvent une dilatation biatriale.
La physiologie restrictive peut ne pas être présente tout au long de l’histoire naturelle, mais seulement à un stade initial (avec une évolution ultérieure vers une phase hypokinétique dilatée). Elle peut également apparaître chez les patients atteints de CMH ou CMD en phase terminale.
La CMR est associée au plus mauvais pronostic de tous les phénotypes de cardiomyopathie. Son pronostic dépend largement de la physiologie restrictive, quelle que soit la cause sous-jacente. Lorsqu’elle est héritée (protéine sarcomérique, la plus prévalente TNNI3), la CMR se présente le plus souvent comme une maladie autosomique dominante et, plus rarement, comme une maladie autosomique récessive ou sporadique.
Prise en charge
Cardiomyopathie spécifique
Maladie d’Anderson-Fabry
Elle est liée à une déficience ou une absence d’une enzyme, l’alpha-galactosidase A (α-GalA), entraînant l’accumulation de produits de dégradation dans la cellule (Lyso Gb3).
Elle est liée au chromosome X ; les hommes sont donc toujours touchés, tandis que l’atteinte d’organes des femmes se développe généralement plus tard dans la vie, mais peut devenir similaire à celle des hommes.
C’est une maladie multisystémique qui affecte en particulier le cœur, les reins et le cerveau.
On distingue deux phénotypes Anderson-Fabry, en fonction du sexe et de la variante génétique pathogène :
– un phénotype clinique sévère, connu sous le nom d’Anderson-Fabry « classique » : apparition de symptômes pendant l’enfance ou l’adolescence suivie d’une défaillance progressive de plusieurs organes, le plus souvent observés chez les hommes (mais pas exclusivement) sans activité enzymatique résiduelle ;
– un phénotype Anderson-Fabry « non classique » ou un phénotype d’apparition plus tardive avec une atteinte systémique incomplète, qui s’observe aussi bien chez les hommes que chez les femmes, avec un certain niveau d’activité enzymatique résiduelle, et qui se manifeste dans la plupart des cas par une atteinte cardiaque isolée.
Quand l’évoquer (tableaux 8 et 9) ?
Elle doit être suspectée chez les patients présentant une HVG et des signes d’alerte cardiaques et extra-cardiaques supplémentaires. L’atteinte cardio-vasculaire se manifeste généralement par une hypertrophie ventriculaire gauche, une fibrose myocardique, une inflammation, une insuffisance cardiaque et des arythmies.
Faire le diagnostic
Le diagnostic est établi par l’évaluation de l’activité α-GalA et la mesure du lyso-Gb3 (produit de dégradation) chez les patients de sexe masculin ; chez les femmes, un test génétique est généralement nécessaire pour confirmer le diagnostic.
Une IRM cardiaque devra être réalisée, voire une biopsie endomyocardique.
Prise en charge
Le traitement enzymatique substitutif est indiqué chez tous les patients symptomatiques atteints de la maladie classique, y compris les enfants, dès les premiers signes d’atteinte d’un organe (car bien moindre efficacité dans les cas avancés de la maladie).
Amylose cardiaque
Il n’est pas dans l’objectif de ce document de fournir des recommandations détaillées sur l’amylose cardiaque. Nous ne rappellerons que les signes évocateurs et la prise en charge diagnostique.
Signes évocateurs d’amylose (figure 14)
Diagnostic d’amylose cardiaque (figure 15)
EN PRATIQUE (figure 16)
• Ces recommandations très complètes insistent donc avant tout sur la mise en évidence rapide d’un phénotype selon lequel une prise en charge ciblée sera réalisée.
• La prise en charge des apparentés doit être aussi une priorité.
• La gestion du risque de mort subite doit se faire tout au long de la maladie.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
