




Publié le 29 fév 2016Lecture 6 min
Hypercholestérolémie familiale : une pathologie insuffisamment reconnue
É. BRUCKERT, Institut hospitalo-universitaire cardio-métabolique, Hôpital de la Pitié-Salpêtrière, Paris
JESFC
L’augmentation du LDL-cholestérol dans le sang est liée à des facteurs génétiques et des facteurs de l’environnement comme la diététique riche en cholestérol ou acides gras saturés, la prise de poids et le vieillissement. La majorité des hypercholestérolémies primaires est appelée polygénique car elles sont la conséquence d’une somme variable d’anomalies génétiques qui quand elles sont isolées ont un impact modeste. Ces formes polygéniques ont donc une sévérité variable en fonction du nombre d’anomalies présentes. Elles ont toutefois comme caractéristique principale l’apparition à l’âge adulte et l’aggravation avec le temps. Les antécédents familiaux de dyslipidémie sont donc aussi très variables (aucun, un ou deux parents avec hypercholestérolémie).
Définition
L’hypercholestérolémie familiale est une entité génétique à part car le risque cardiovasculaire en l’absence de traitement est multiplié par 13. Elle est par définition une maladie monogénique autosomique dominante.
Son diagnostic repose sur 3 caractéristiques fondamentales :
Une élévation du LDL-cholestérol. Dans la forme familiale le niveau de cholestérol dans le sang est en moyenne le double de la normale. Dans les autres formes (parfois appelées polygéniques), le niveau est le plus souvent plus bas que dans la forme familiale. Un Français sur 500 a une hypercholestérolémie familiale alors qu’on peut considérer qu’au moins un tiers des Français a une hypercholestérolémie polygénique (mélange de prédisposition génétique, de diététique non adaptée ou parfois de surpoids). L’élévation du LDL-C existe depuis la naissance.
Une histoire familiale compatible avec une forme autosomique dominante. Dans cette histoire, il existe souvent une maladie cardiovasculaire précoce.
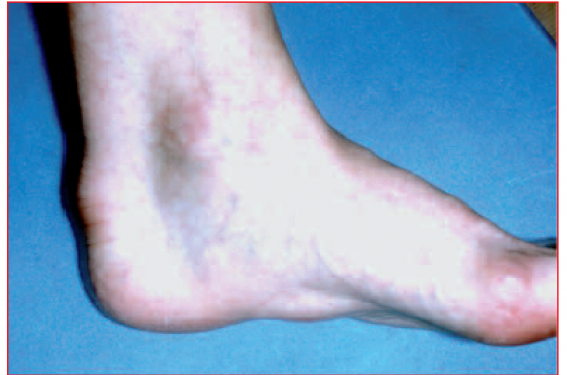
Des signes cliniques. Les dépôts extravasculaires de cholestérol sont fréquents et comprennent l’arc cornéen (évoque une hypercholestérolémie familiale quand il est présent avant 45 ans), le xanthélasma, les xanthomes tendineux (figure). Ils sont le plus souvent visibles et palpables sur les tendons d’Achille et sur les tendons extenseurs des doigts de la main. Les xanthomes tendineux sont pratiquement pathognomoniques de forme familiale.
Figure. Xanthomes tendineux.
Le diagnostic qui peut se faire sur un score clinique et biologique (échelle développée en Hollande, tableau) peut aussi être fait ou confirmé par l’analyse génétique (cf. infra).
L’étude de Copenhague faite dans une grande population démontre que les patients atteints de forme familiale ont un risque de maladie coronaire multiplié par 13. D’autres études confirment ce risque très élevé(revue in 1). De façon intéressante, une vaste étude génétique montre que chez les sujets ayant eu un infarctus du myocarde il y a 13 fois plus de mutation dite disruptive du gène du récepteur aux LDL(2).
L’hypercholestérolémie familiale doit être diagnostiquée car sa prise en charge est spécifique
La première raison justifiant la nécessité d’identifier clairement ces patients est que le risque de faire un accident cardiovasculaire jeune est très élevé et que le traitement correctement conduit protège contre ce risque.
La deuxième raison est qu’il faut identifier les autres membres de la famille quand un individu est diagnostiqué avec cette forme familiale. Les enfants doivent être dépistés à partir de l’âge de 3 ans et traités à partir de l’âge de 10 ans. Enfin la dernière raison est que l’identification et le diagnostic génétique quand il est fait conduisent le plus souvent les patients à une meilleure observance des traitements.
Les anomalies génétiques responsables d’hypercholestérolémie familiale touchent le plus souvent le récepteur des LDL. En France, les mutations sur ce récepteur expliquent 80 % des formes familiales. Il existe plus de 1 000 mutations décrites. Certaines entraînent une absence totale du récepteur ou de sa fonction et s’accompagnent d’une forte élévation du cholestérol. D’autres laissent persister une petite partie de la fonction de la protéine et sont un peu moins sévères. La mutation qui touche l’apolipoprotéine B est plus rare et touche un faible pourcentage de patients. Les mutations qui touchent la protéine PCSK9 sont très rares avec seulement quelques familles touchées. Il reste des formes familiales typiques pour lesquelles la mutation n’est pas identifiée. Le diagnostic génétique est possible dans les laboratoires agréés. Il en existe 4 en France.
La prise en charge de l’hypercholestérolémie familiale
La prise en charge d’une hypercholestérolémie familiale comporte systématiquement deux catégories de mesures :
- des mesures dites hygiénodiététiques associées au traitement des autres facteurs de risque quand ils existent ;
- des traitements médicamenteux.
Mesures hygiéno-diététiques
Chez l’adulte, les mesures diététiques recommandées sont essentiellement :
- une réduction des apports globaux en matières grasses ;
- avec essentiellement une réduction de l’apport en graisses saturées ;
- et de façon conjointe une réduction des aliments riches en cholestérol.
Il est donc conseillé :
- de réduire la consommation de graisses d’origine animale (charcuteries, viandes grasses, beurre, crème fraîche, lait et laitages non écrémés, fromages gras, etc.) ;
- d’éviter les aliments les plus riches en cholestérol tels que les abats, et de limiter la consommation des œufs à 2-3 par semaine ;
- de privilégier les matières grasses d’origine végétale : huiles d’olive et d’arachide, huiles de tournesol, de maïs, de pépins de raisin, huiles de colza, de soja, de noix ;
- d’encourager la consommation de viandes maigres, de volailles, de poissons, de lait et de laitages écrémés ou partiellement écrémés, ainsi que la consommation de fruits et légumes ;
- et d’éviter les aliments d’origine industrielle riches en mauvaises graisses, tels que viennoiseries, pâtisseries, certains biscuits, plats cuisinés, etc.
De plus, il est recommandé d’incorporer dans l’alimentation un apport de fibres, ainsi qu’un apport de stérols ou de stanols végétaux.
Traitements médicamenteux
Dans l’hypercholestérolémie familiale, la classe la plus importante de médicaments pour réduire le LDL-cholestérol est la famille des statines. Plusieurs molécules sont commercialisées qui permettent d’obtenir une réduction du LDL-cholestérol allant de 30 à 50 %, diminution qui dépend à la fois de la molécule choisie et de la dose utilisée. En pratique, compte tenu de l’élévation importante du LDL-C, il est le plus souvent nécessaire de recourir aux statines les plus puissantes en particulier l’atorvastatine ou la rosuvastatine.
Avant l’utilisation des statines, il était classique d’utiliser des résines qui sont des substances qui se lient dans l’intestin à des acides biliaires. Ce traitement (colestyramine) est associé à de nombreux effets secondaires digestifs, ce qui limite l’acceptabilité pour les patients. La réduction du LDL-cholestérol obtenue avec les résines, est de 15 à 25 % en fonction de la dose utilisée. L’ézétimibe est une autre molécule qui agit au niveau intestinal, mais par un mécanisme différent.
L’ézétimibe bloque de façon spécifique l’absorption intestinale du cholestérol qu’il soit d’origine alimentaire ou biliaire. La réduction moyenne du LDL-cholestérol observée sous ézétimibe est de l’ordre de 20 %. L’ézétimibe doit essentiellement être utilisé en association avec une statine lorsque le traitement par statine ne permet pas d’abaisser suffisamment le taux de LDL-cholestérol.
Chez l’enfant et l’adolescent, il est actuellement recommandé de proposer un traitement médicamenteux à partir de l’âge de 8 à 10 ans lorsque le taux de LDL-cholestérol reste supérieur à 1,90 g/l, après une période d’au moins 6 mois de mesures hygiéno-diététiques. Il est maintenant recommandé de choisir comme traitement de première intention chez l’enfant et l’adolescent une statine, comme chez l’adulte, mais en utilisant la dose efficace la plus faible(3). Un traitement plus précoce peut parfois être justifié dans des formes sévères, en général après avis spécialisé.
Dans la forme particulièrement grave de l’hypercholestérolémie familiale, forme dite homozygote (quand les deux parents ont transmis la maladie), il est possible de proposer la LDL-aphérèse.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
