



Publié le 13 fév 2018Lecture 11 min
La sclérodermie systémique
Paul LEGENDRE(1), Luc MOUTHON(1,2) (1)Service de Médecine Interne, Hôpital Cochin, Centre de Référence National pour les Maladies Systémiques Auto-immunes Rares, Paris (2)Institut Cochin, INSERM U1016, CNRS UMR 8104, Université Paris-Descartes, Paris
La sclérodermie systémique est une pathologie rare associant une accumulation de matrice extracellulaire, une atteinte microvasculaire oblitérante et des anomalies de l’immunité humorale et cellulaire.
Elle est caractérisée par des lésions de fibrose (sclérose cutanée et fibrose pulmonaire) et des anomalies vasculaires (phénomène de Raynaud, hypertension artérielle pulmonaire [HTAP]).
Sa prise en charge repose sur le traitement symptomatique des atteintes les plus fréquentes et des complications viscérales (HTAP, crise rénale).
Un traitement immunosuppresseur est nécessaire à la phase initiale de la maladie en cas d’atteinte cutanée diffuse et/ou de pneumopathie interstitielle aggravative.
Généralités
La sclérodermie systémique (ScS) est une connectivite rare, touchant principalement les femmes et potentiellement fatale. La ScS est une pathologie rare du tissu conjonctif caractérisée par un durcissement (sclérose) de la peau avec un sex-ratio entre 3/1 et 8/1. Sa prévalence varie de 30 (Japon) à 240 (Amérique du Nord et Australie) par million d’habitants(1).
Les manifestations cliniques de la ScS associent des lésions de fibrose (sclérose cutanée et fibrose pulmonaire), des anomalies vasculaires (phénomène de Raynaud [PR], hypertension artérielle pulmonaire [HTAP]) et des stigmates d’auto-immunité (auto-anticorps dans le sérum).
Les patients atteints de ScS ont un risque de décès 3,5 fois plus élevé que les sujets sains du même âge, ce qui fait de la ScS la connectivite ayant le moins bon pronostic(2). Le pronostic est d’autant plus péjoratif que le patient a une forme cutanée diffuse, fréquemment associée à des atteintes viscérales : atteinte interstitielle pulmonaire, digestive basse, rénale ou cardiaque.
Physiopathologie
Au cours de la ScS, il existe une interaction anormale entre les fibroblastes, les cellules endothéliales et les cellules de l’immunité. On observe une production exagérée de matrice extracellulaire par les fibroblastes via la sécrétion de facteurs pro-fibrosants : transforming growth factor β (TGF-β) et platelet derived growth factor (PDGF). De plus, les vaisseaux sont le siège d’un remodelage intense : on observe un excès d’apoptose des cellules endothéliales et une vasoconstriction intense liée à la production d’endothéline-1 (ET-1).
En outre, au cours de la ScS, la présence d’auto-anticorps dans le sérum des patients témoigne d’une dérégulation des lymphocytes B.
De nombreux facteurs environnementaux ont été reconnus comme impliqués dans la survenue d’une ScS, comme l’exposition à la silice, aux solvants et/ou aux hydrocarbures.
Enfin, la part génétique reste très modeste dans la pathogénie de la ScS.
Diagnostic
De nouveaux critères diagnostiques de l’American College of Rheumatology (ACR) et de l’European League Against Rheumatism (EULAR) ont été établis en 2013(3).
Ils se basent sur des items associés à des points, où l’obtention d’un score de 9 permet de classer le patient comme ayant une ScS. La sclérose cutanée remontant au-dessus des articulations métacarpo-phalangiennes suffit à poser le diagnostic, sous réserve que les lésions aient débuté en distalité et qu’il ne s’agisse pas d’une pseudo-sclérodermie. En son absence, d’autres items doivent être associés pour affirmer le diagnostic (tableau).
On continue néanmoins à distinguer, en fonction de l’extension de l’atteinte cutanée, les formes diffuses, remontant au-dessus des coudes et des genoux, ou limitées, intéressant la distalité des membres et le visage en épargnant les zones proximales des membres et le tronc.
ScS : sclérodermie systémique.
* Ces critères sont applicables à tout patient pouvant être inclus dans une étude sur la ScS. Ils ne sont pas applicables à des patients qui ont des épaississements cutanés épargnant les doigts ou à des patients ayant une pseudo-sclérodermie pouvant mieux expliquer leurs manifestations (par exemple fibrose néphrogénique, morphée généralisée, fasciite à éosinophiles, sclérose diabétique, scléromyxoedème, érythromyalgie, porphyrie, lichen scléreux, réaction du greffon contre l’hôte, cheiro-arthropathie diabétique).
† Le score total est déterminé par l’addition des scores maximaux dans chaque catégorie. Les patients totalisant un score > 9 sont classés comme ayant une ScS.
Manifestations de la sclérodermie systémique
Les manifestations cliniques de la ScS associent des lésions de fibrose, des anomalies vasculaires et des stigmates d’autoimmunité.
Pathologie vasculaire
Le phénomène de Raynaud (PR) est extrêmement fréquent au cours de la ScS, il touche les extrémités selon une crise typique à 3 phases : pâleur syncopale avec anesthésie limitée à quelques doigts (figure 1B), phase de cyanose paresthésique (figure 1C), phase érythémateuse tardive douloureuse. Lorsqu’il est sévère, il peut se compliquer d’ulcérations digitales (UD) (figure 2B) et de lésions ischémiques chroniques à risque de surinfection.
L’HTAP est définie par une pression artérielle pulmonaire moyenne > 25 mmHg mesurée par cathétérisme droit. Sa prévalence varie de 7,85 % à 13 % des patients sclérodermiques. L’absence de signe clinique spécifique de l’HTAP entraîne un retard diagnostique d’autant plus qu’elle est associée à un mauvais pronostic (survie de 55 % à un an)(4). Les télangiectasies (figure 2A) sont plus nombreuses chez les patients qui développent une HTAP et reflètent elles aussi la microangiopathie sclérodermique. Chez 2 à 5 % des patients sclérodermiques, une crise rénale sclérodermique (CRS) peut survenir. Elle se manifeste par une HTA maligne et une insuffisance rénale aiguë oligurique. Elle demeure une importante cause de mortalité (de 18 à 36 % au cours de la première année)(5), bien que son pronostic ait été amélioré par l’utilisation des inhibiteurs de l’enzyme de conversion (IEC).
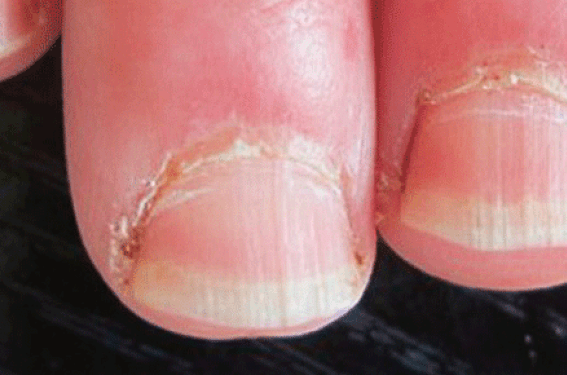
Figure 1. Sclérodermie systémique. 1A : Hypertrophie des cuticules. 1B : Phénomène de Raynaud (phase syncopale) localisée au 3e doigt de la main droite. 1C : Phénomène de Raynaud (phase cyanique) de la main droite.
Figure 2. Sclérodermie systémique. 2A. Télangiectasies. 2B. Ulcère digital surinfecté du 4e doigt de la main gauche. 2C. Acro-ostéolyse des 2e et 3e rayons de la main gauche, dépigmentation en regard des métacarpophalangiennes. 2D. Calcinoses multiples de la main droite.
Pathologie fibrosante
La sclérose cutanée peut évoluer en 3 phases :
– une phase œdémateuse inconstante, observée surtout dans les formes diffuses, caractérisée par un gonflement des doigts et des mains qui sont infiltrés, donnant un aspect boudiné ;
– une phase indurée, durant laquelle apparaissent les lésions de sclérose cutanée, pouvant aller d’une sclérodactylie modérée à un épaississement cutané très marqué, la peau n’étant quelquefois plus plissable, pouvant adhérer aux plans profonds et conduisant progressivement à la rétractation irréductible des doigts en flexion à l’origine du signe de la prière. La sclérose du visage est responsable d’une disparition des rides du front, d’un nez aquilin, d’une diminution de l’ouverture buccale (microstomie) mesurée par la distance entre les arcades dentaires (< 40 mm) ;
– une phase atrophique, au cours de laquelle on note une atrophie puis une disparition de l’hypoderme aboutissant à une peau fine, des lèvres fines et une accentuation des plis radiés péribuccaux.
Les atteintes cutanées se compliquent souvent de troubles de la pigmentation qui peuvent associer des plages de dépigmentation (figure 2C) ou d’hyperpigmentation.
L’évaluation de la sclérose cutanée se fait par la mesure du score de Rodnan modifié (mRSS). Ce score évalue, en 17 zones du corps, par la simple palpationcutanée, l’importance de l’épaississement : 0 : épaisseur cutanée normale ; 1 : épaississement minime ; 2 : épaississement modéré ; 3 : épaississement majeur, avec adhérence aux plans profonds. Les calcinoses correspondent au dépôt de cristaux d’hydroxyapatite dans les tissus sous-cutanés (figure 2D).
Le poumon est, lui aussi, le siège de phénomènes de fibrose. La prévalence des pneumopathies interstitielles diffuses (PID) varie de 16 à 100 % au cours de la ScS. Elle survient plus fréquemment dans les formes diffuses de la maladie, en présence d’anticorps anti-Scl70 et/ou dans les premières années d’évolution de la maladie. Sa progression étant peu symptomatique, elle est mieux évaluée par la diminution de la capacité vitale forcée (CVF). Par ailleurs, ces patients peuvent développer une hypertension pulmonaire secondaire à la PID.
L’atteinte du tube digestif est très fréquente au cours de la ScS. Le RGO est la manifestation digestive la plus précoce et la plus fréquente de la maladie, présente dans 80 % des cas environ. Des troubles de motilité surviennent à tous les étages du tube digestif (dysphagie, gastroparésie, ralentissement du transit du grêle pouvant aboutir à un tableau de pseudoocclusion), atteinte ano-rectale responsable d’une incontinence.
Autres atteintes
Les at teintes musculosquelettiques se manifestent par des douleurs articulaires et musculaires, des frictions tendineuses et des limitations de la mobilité articulaire impactant énormément la qualité de vie des patients.
Des arthralgies et une raideur des doigts, mains et poignets sont fréquentes chez les patients sclérodermiques, mais des synovites sont possibles.
L’atteinte myocardique est la résultante d’anomalies micro-circulatoires et d’un remodelage tissulaire excessif. Elle entraîne des troubles du rythme et de la conduction et/ou une péricardite.
Examens complémentaires
Ils permettent de confirmer le diagnostic de ScS, de dépister les complications et d’assurer le suivi évolutif (encadré ci-dessous).
CPK : créatine phophokinase ; DLCO : diffusion libre du monoxyde de carbone ;
ECG : électrocardiogramme ; EFR : épreuves fonctionnelles respiratoires ; LDH : lactates déshydrogénases ; NT-pro BNP : N-Terminal pro-brain natriuretic peptide (fraction N-terminale du peptide natriurétique de type B), VIT : vitesse d’insuffisance tricuspidienne
La confirmation du diagnostic de ScS nécessite une capillaroscopie péri-unguéale à la recherche de mégacapillaires associés à des plages avasculaires et la mise en évidence des anticorps antinucléaires de spécificité anticentromères/antitopoisomérase 1(Scl70) ou anti-ARN polymérase III en leur absence. Le dépistage des atteintes d’organes comprend une échographie cardiaque transthoracique avec mesure de la vitesse d’insuffisance tricuspide (VIT), un scanner thoracique en coupes fines haute résolution (qui peut révéler des aspects en verre dépoli, des réticulations intralobulaires ou des aspects en rayon de miel) et des épreuves fonctionnelles respiratoires (EFR) avec mesure de la diffusion libre du monoxyde de carbone (DLCO), des radiographies des mains à la recherche de lésions de calcinose et éventuellement une manométrie œsophagienne pour préciser l’atteinte œsophagienne si le tableau est atypique par ailleurs.
Lorsque le diagnostic de ScS est établi, en plus des examens précédents, il faut réaliser une numération formule sanguine (NFS) pour rechercher une anémie, un frottis sanguin, un dosage de l’haptoglobine, des dosages de lactico-déshydrogénases (LDH) à la recherche d’une hémolyse, un bilan hépatique complet (transaminases, phosphatases alcalines, gamma GT, bilirubine libre et conjuguée), un dosage de créatine phophokinase (CPK), une électrophorèse des protéines plasmatiques, un ionogramme plasmatique, créatininémie, urémie, calcémie, phosphorémie, un dosage du NT-pro BNP et une étude du sédiment urinaire. Le bilan biologique sera assorti d’une recherche d’auto-anticorps pouvant être associés à diverses manifestations cliniques de la ScS, un ECG, une échocardiographie. D’autres auto-anticorps, comme les anti-Pm/Scl, antifibrillarine, anti-MMP 1 et 3, anti-PDGFR et anti-Nag-2, peuvent également être identifiés au cours de la ScS. Cependant, seuls les deux premiers peuvent être recherchés. On réalisera également une fibroscopie œsogastrique s’il y a un point d’appel, à la recherche d’une œsophagite, voire d’une sténose peptique.
Enfin, on pratiquera annuellement de manière systématique un bilan biologique similaire à celui du diagnostic, associé à des EFR avec DLCO et à une échocardiographie transthoracique.
Traitement
La prise en charge des patients sclérodermiques se fait selon deux axes : le traitement des complications viscérales et le traitement de fond.
Traitement des complications viscérales
La prise en charge du phénomène de Raynaud nécessite avant tout le respect de mesures préventives : protection contre le froid et l’humidité, arrêt du tabac et éviter certains traitements aggravants (bêtabloquants, antimigraineux, collutoires contenant des vasoconstricteurs comme la phényléphrine). Un traitement inhibiteur calcique est indispensable chez la plupart des patients. L’iloprost intraveineux est prescrit en cas de survenue de troubles trophiques distaux. Un inhibiteur des récepteurs de l’endothéline est nécessaire en prévention de la survenue de nouveaux ulcères digitaux chez les patients ayant une maladie ulcéreuse digitale sévère.
Les traitements spécifiques de l’HTAP seront prescrits après réalisation d’un cathétérisme droit confirmant le diagnostic d’HTAP. Chez les patients ayant une dyspnée de stade II ou III, on privilégiera les traitements oraux, antagonistes des récepteurs de l’ET-1 (bosentan, ambrisentan, macitentan) et inhibiteurs de la phosphodiestérase de type 5 (sildénafil, tadalafil) qui peuvent être éventuellement combinés. Les inhibiteurs de la guanylate cyclase (riociguat) ont également une efficacité démontrée. Un agoniste des récepteurs de la prostacycline vient d’obtenir également une autorisation de mise sur le marché dans l’HTAP. Chez les patients ayant une dyspnée de stade IV, on privilégiera un traitement par analogues de la prostacycline (époprosténol, tréprostinil), éventuellement en association avec un traitement oral. En cas d’échec du traitement médical, la transplantation bipulmonaire ou cardiopulmonaire constitue la solution thérapeutique ultime chez les patients ne présentant pas de contre-indication.
Les IEC, comme le captopril ou l’énalapril, ont permis d’améliorer la survie au décours de la crise rénale sclérodermique (CRS). Ils n’ont en revanche pas de place en prévention primaire.
En plus des mesures posturales pour limiter le RGO et la survenue d’une œsophagite, l’utilisation d’inhibiteurs de la pompe à protons (oméprazole ou lansoprazole) fait l’objet d’un consensus. Les agents prokinétiques comme la dompéridone peuvent être prescrits chez les patients ayant un reflux persistant. Cependant, il est important de mentionner que la prise doit s’effectuer au moins 30 minutes avant le repas.
En cas de gastroparésie, l’érythromycine et l’octréotide (analogue de la somatostatine) ont un effet prokinétique respectivement sur l’estomac et le grêle. Un traitement antibiotique est proposé en cas de pullulation microbienne. L’objectif principal est d’éviter la dénutrition.
Les arthromyalgies sont sensibles aux antalgiques, aux antiinflammatoires non stéroïdiens, voire à la corticothérapie à faible dose. Chez les patients ayant une polyarthrite, un traitement par méthotrexate peut être proposé.
Traitement de fond
La corticothérapie à faible dose peut apporter un bénéfice sur les manifestations articulaires, musculaires, sur les pneumopathies infiltrantes diffuses (PID) évolutives, voire sur les formes œdémateuses de ScS. Néanmoins, le risque accru de CRS limite l’utilisation des corticoïdes et la dose maximale de 15 mg/j ne doit pas être dépassée. Les traitements immunosuppresseurs et immunomodulateurs doivent être réservés aux ScS diffuses de diagnostic récent (moins de 3 à 5 ans) ou évolutives.
Le méthotrexate est recommandé par l’EULAR comme traitement des formes précoces de ScS cutanées diffuses à la dose de 0,3 mg/kg par semaine (voie orale ou sous-cutanée).
Le mycophénolate mofétil semble avoir un bénéfice dans l’atteinte cutanée diffuse(6).
L’EULAR recommande l’utilisation du cyclophosphamide par voie intraveineuse (6 bolus à la dose de 600 mg/m2 à 4 semaines d’intervalle) dans la prise en charge de la PID, avec un relais par mycophénolate mofétil ou azathioprine(6).
L’abatacept, le rituximab et le tocilizumab semblent tous les trois pouvoir apporter un bénéfice dans les formes diffuses de ScS ou dans le traitement des PID aggravatives, sans toutefois que leur utilisation puisse être recommandée en première intention. L’autogreffe de cellules souches périphériques, précédée d’un conditionnement par cyclophosphamide, sérum antilymphocytaire et corticoïdes peut être justifiée chez des patients ayant une forme diffuse rapidement progressive et/ou avec atteinte viscérale aggravative(7).
Autres traitements
Un anticorps monoclonal ciblant le transforming growth factor (TGFß), le fresolimumab, a permis dans une étude pilote sur un petit collectif de patients, une amélioration du score cutané et une régression de l’infiltration de la peau par les myofibroblastes. L’interféron-α (IFN-α ), la relaxine recombinante, l’infliximab et l’imatinib n’offrent pas de bénéfice démontré au cours de la ScS(6).
Par ailleurs, les mesures non pharmacologiques comme l’arrêt du tabac, la rééducation fonctionnelle et l’éviction de substances telles que la silice ou les solvants organiques permettent une amélioration de la fonction respiratoire et de la qualité de vie.
"Publié dans Dermatologie Pratique"
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
