



Publié le 20 nov 2007Lecture 9 min
L'athérosclérose : une maladie inflammatoire
H. AIT-OUFELLA, A. TEDGUI et Z. MALLAT, Centre de Recherche Cardiovasculaire Inserm Lariboisière, Paris
L’athérosclérose est une maladie inflammatoire chronique des gros vaisseaux. Le processus de formation de la plaque fait intervenir plusieurs acteurs pathogènes de l’inflammation, en particulier les monocytes/macrophages, les lymphocytes T CD4+ de type Th1 et potentiellement de type Th2. Récemment nous avons identifié le rôle majeur d’une nouvelle sous-population de lymphocytes T CD4+, appelés T régulateurs (Tregs), dans la protection contre le développement de l’athérosclérose. Plusieurs études expérimentales ont montré que l’expansion in vivo de ces Tregs est associée à une diminution du développement et/ou de la progression des plaques d’athérosclérose chez la souris, ouvrant le champ vers de nouvelles thérapeutiques immunomodulatrices des maladies cardiovasculaires.
Les maladies cardiovasculaires sont aujourd’hui un véritable problème de santé publique puisqu’elles constituent une des premières causes de mortalité dans le monde. Selon l’OMS, 7,2 millions de décès par an dans le monde sont liés à des cardiopathies ischémiques et 4,6 millions de décès à des accidents vasculaires cérébraux. L’athérosclérose est la principale étiologie des maladies cardiovasculaires. Elle provoque des pathologies chroniques comme l’angor d’effort ou la claudication à la marche et des maladies aiguës plus bruyantes et potentiellement mortelles comme l’infarctus du myocarde et l’accident vasculaire cérébral.
Les travaux épidémiologiques et anatomopathologiques réalisés chez l’homme, associés aux études in vitro, à partir de cellules humaines, ont apporté la preuve que l’athérosclérose est une maladie inflammatoire chronique des artères de gros et moyen calibre(1). Les modèles expérimentaux animaux, essentiellement murins, ont été très utiles et complémentaires pour disséquer les mécanismes physiopathologiques qui aboutissent à la formation de la plaque d’athérosclérose. Ainsi, grâce à ces différents outils, il a été montré que la formation de la plaque d’athérosclérose comporte plusieurs étapes :
• l’activation de l’endothélium artériel par les LDL oxydés,
• l’attraction puis la diapédèse des monocytes et des lymphocytes T circulants dans l’intima,
• la production de cytokines pro- et anti-inflammatoires,
• la production de protéases matricielles,
• enfin, l’induction d’apoptose des différents types cellulaires aboutissant à la formation d’un noyau lipidique nécrotique.
Activation endothéliale
L’infiltration et la rétention des lipoprotéines de basse densité (LDL) puis leur oxydation dans l’espace sous-endothélial active les cellules endothéliales(2). Le site privilégié de cette activation correspond aux zones de bifurcation artérielles, là où les forces de cisaillement sont les plus faibles et les flux turbulents les plus importants. Ainsi, les facteurs chimiques et les facteurs physiques agissent de concert pour activer l’endothélium et initier la formation de la plaque.
Les cellules endothéliales, une fois activées, expriment plusieurs types de molécules d’adhérence leucocytaire et de chimiokines qui permettent aux cellules sanguines de rouler puis d’adhérer à la surface vasculaire. P-sélectine, VCAM-1 et IL-8(3,4) sont les plus importantes à ce stade. Ensuite, des chimiokines telles que monocyte, chemokine protein (MCP-1), produites par les cellules endothéliales et les cellules musculaires, stimulent la migration des cellules inflammatoires vers le sous-endothélium. L’importance et le rôle des protéines précédemment citées résultent d’études animales qui ont montré que leur blocage pharmacologique ou leur délétion génétique induit une diminution significative de la taille des lésions d’athérosclérose(3,4).
Rôle des monocytes/macrophages
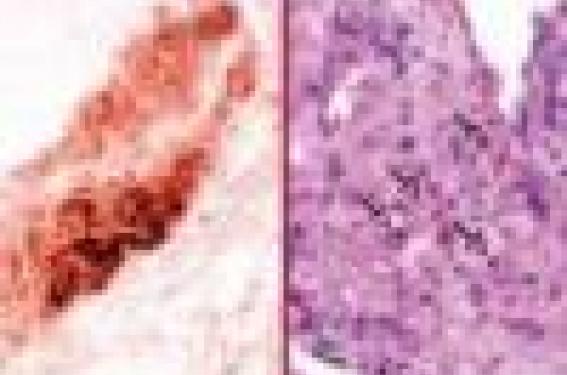
Le rôle fondamental des monocytes/macrophages dans le développement de la plaque d’athérosclérose est évoqué depuis longtemps car les lésions athéroscléreuses sont très riches en macrophages, surtout aux stades précoces (figure 1A). Les études animales ont confirmé ce point. Les souris déficientes en M-CSF5, un facteur de croissance indispensable à la prolifération et/ou la maturation des monocytes circulants en macrophages, sont résistantes au développement de l’athérosclérose, malgré des taux de cholestérol circulants très élevés, suggérant le rôle majeur des macrophages dans le développement des lésions. Les macrophages, via certains récepteurs « éboueurs » (SR-AI, SR-AII, CD36…), phagocytent des LDL oxydés, des phospholipides, des débris cellulaires et deviennent des cellules spumeuses. Ils entretiennent le processus inflammatoire en produisant des cytokines pro-inflammatoires comme l’IL-12 (qui activent les lymphocytes T CD4+ pathogènes) et contribuent à déstabiliser la plaque en sécrétant, entre autres, des radicaux libres oxygénés et des métalloprotéases matricielles(6).
Ces cellules inflammatoires jouent également un rôle central dans la vulnérabilité des plaques d’athérosclérose. Cette conclusion résulte des travaux anatomopathologiques réalisés notamment par l’équipe de Davis et Virmani qui ont comparé la composition des plaques coronaires rompues à celle de plaques stables prélevées sur des cadavres humains. Ces études ont montré que les plaques rompues, responsables d’événements ischémiques, sont caractérisées par une infiltration importante de cellules inflammatoires, particulièrement les macrophages, et une diminution d’éléments stabilisants comme les cellules musculaires lisses et le collagène(7, 8).
Rôle des lymphocytes T
Rôle pathogène des lymphocytes T CD4+
Il a fallu attendre quelques années pour montrer que les lymphocytes T CD4+ ont aussi un rôle pathogène dans l’athérosclérose. Tout d’abord, ils sont présents dans les plaques (figure 1b), surtout au stade précoce où ils représentent 10 à 20 % de la population cellulaire totale. Ensuite, ils sont activés car ils se trouvent fréquemment colocalisés en immunohistochimie avec le complexe majeur d’histocompatibilité de classe II. Chez la souris apoE-/-, sensible à l’athérosclérose, la déficience en lymphocytes T et B induite par croisement avec des souris SCID-/- provoque une diminution de 90 % de la taille des lésions au niveau du sinus aortique, en présence de taux de cholestérol circulant inférieurs à 6 g/l. De plus, la reconstitution de la population lymphocytaire T, par injection intraveineuse de lymphocytes CD4+, augmente considérablement les plaques aussi bien au niveau de la crosse de l’aorte que du sinus aortique(9).
Figure 1. Détection par immunohistochimie des macrophages en brun (A) et des lymphocytes T en rouge (B) au sein de plaque d’athérosclérose isolées de souris apoE-/-.
Les lymphocytes T de type Th1 sont proathérogènes
Les lymphocytes T impliqués dans l’athérosclérose sont essentiellement de type Th1 produisant de grandes quantités d’interféron gamma (IFNg), contrairement aux lymphocytes T de type Th2 qui produisent de l’interleukine-4 (IL-4), de l’IL-5, et de l’IL-10. L’IFNg est retrouvé en immunohistochimie dans les plaques d’athérosclérose dans les mêmes régions que les cellules T. Le transfert in vivo de lymphocytes T CD4+, réalisé dans le travail précédemment décrit, s’accompagne d’une augmentation considérable des taux plasmatique et splénique d’IFNg(9). Le blocage de la signalisation de l’IFNg dans les lymphocytes T, obtenu par sa délétion(10) ou la délétion de son récepteur(11), induit une diminution de la taille des plaques d’athérosclérose et un phénotype lésionnel plus stable.
L’IL-18 est aussi une cytokine pro-athérogène qui stimule la production lymphocytaire d’IFNg, en synergie avec l’IL-12. L’injection d’IL-18 recombinante chez la souris s’accompagne d’une augmentation de la taille des plaques d’athérosclérose avec une augmentation de la production systémique d’IFNg(12). Dans le laboratoire, nous avons montré que le blocage in vivo de l’IL-18 par son inhibiteur physiologique l’IL-18 BP (binding protein) permet une diminution de la taille des lésions tout en induisant un phénotype lésionnel stable(13). Chez l’homme, l’IL-18 est exprimée dans les plaques athéroscléreuses et son taux au sein des lésions vasculaires (évalué en PCR quantitative) est plus important chez les patients symptomatiques (AIT ou AVC) que chez les patients asymptomatiques(14). Enfin, il a été montré sur une cohorte de plus de 1 000 patients que le taux d’IL-18 plasmatique est, chez les coronariens, un facteur pronostique d’événements cardiovasculaires(15).
La voie Th2 est potentiellement pro-athérogène
Les lymphocytes de type Th2, producteurs d’IL-4, ont été d’abord considérés comme protecteurs contre le développement de l’athérosclérose. Cependant, plusieurs travaux récents remettent en cause cette hypothèse. Chez l’animal, lorsque l’on invalide le gène codant pour l’IL-4, on diminue la taille des plaques au niveau du sinus aortique et de la crosse aortique(16). Les études épidémiologiques chez l’homme ont montré que le risque cardiovasculaire des patients souffrant d’asthme, une maladie de type Th2, est nettement supérieur à celui des patients non asthmatiques. Dès lors, la théorie qui oppose la voie Th1 à la voie Th2 comme le « bien et le mal » dans l’athérogenèse est devenue criticable.
Un sous-type de lymphocytes T protecteurs contre l’athérosclérose
Dans le laboratoire, nous avons émis l’hypothèse que, dans le contexte de l’athérosclérose, la dysfonction lymphocytaire T est liée à un défaut de régulation par des cellules T régulatrices qui contrôlent la réponse immuno-inflammatoire(17). Cette théorie est d’autant plus séduisante que la génération et le mode d’action des lymphocytes T régulateurs (Tregs) font intervenir l’IL-10(18,19) et le transforming growth factor b (TGFb)(20,21), deux cytokines clairement identifiées comme anti-inflammatoires et anti-athérogènes par différentes équipes. En utilisant plusieurs modèles murins nous avons montré que la déplétion en Tregs s’accompagne d’une accélération de la maladie athéroscléreuse et que la reconstitution d’un pool physiologique de ces cellules empêche l’accélération de la maladie (figure 2). Enfin, nous avons montré que l’injection de Tregs stabilise les plaques avec des lésions moins riches en cellules inflammatoires (macrophages) et plus riches en collagène, un composant stabilisant de la plaque(22, 23).
Ces travaux commencent à trouver une relevance chez l’homme puisqu’il a été récemment montré que les Tregs sont diminués quantitativement et qualitativement chez les patients coronariens instables par rapport aux patients avec angor stable(24).
Figure 2. Le transfert de splénocytes déficients en Tregs induit une augmentation des plaques d’athérosclérose au niveau du sinus aortique (B), la reconstitution d’un pool physiologique en Tregs prévient cette augmentation (C). D’après Ait-Oufella et al, Nat Med 2006.
Perspectives thérapeutiques
L’identification récente de cette nouvelle population protectrice dans l’athérosclérose a ouvert la voie à différentes approches thérapeutiques, dont le but commun est d’expandre in vivo des Tregs pour limiter le développement de la maladie vasculaire. Une équipe française a récemment montré dans un travail publié dans le New England Journal of Medicine que l’injection d’anticorps anti-CD3, qui induit une augmentation des Tregs et leur activité dépendante du TGFb, prévenait l’apparition du diabète chez des jeunes adultes prédiabétiques(25). Cette stratégie immunomodulatrice a été testée dans l’athérosclérose expérimentale et a montré des résultats prometteurs puisque l’injection d’anti-CD3 a été capable de retarder l’apparition des lésions artérielles et de retarder leur progression. Cet effet macroscopique était bien associé à une augmentation du facteur de transcription des Tregs dans les ganglions périphériques et la rate(26).
Une autre stratégie, appelée induction de tolérance, consiste à expandre des Tregs par la prise répétée d’un antigène donné à faibles doses, en l’absence d’adjuvant. Une équipe hollandaise a récemment montré que l’administration orale de faibles doses de LDL oxydés inhibe l’initiation et la progression de l’athérosclérose chez la souris avec une augmentation de la production de TGFb (antiathérogène) et une augmentation des Tregs dans la rate et les ganglions mésentériques(27). À travers ce dernier travail expérimental apparaît l’idée d’une induction de réponse protectrice « intelligente », active en réponse à un antigène donné, en l’occurrence les LDL oxydés, le principal antigène impliqué dans la réponse inflammatoire au sein de la plaque, afin de cibler spécifiquement la maladie et de réduire au maximum le développement d’une immunosuppression systémique. La finalité est d’exploiter les propriétés protectrices de ces Tregs de façon ciblée en s’affranchissant d’éventuels effets secondaires systémiques. D’ailleurs, dans le laboratoire nous développons actuellement une stratégie d’induction de tolérance plus ciblée en utilisant des fractions peptidiques des LDL oxydés, identifiées comme les parties les plus immunogènes chez des patients coronariens instables.
En pratique
L'athérosclérose est une maladie chronique des artères de gros et moyen calibres. Ces dernières années ont vu émerger le rôle fondamental de l'inflammation dans la genèse de la maladie et de ses complications. À côté de l'immunité innée incarnée par les macrophages, s'est dessiné le rôle complexe de l'immunité adaptative; tantôt pathogène pour les lymphocytes de type Th1, tantôt protecteur pour les lymphocytes T régulateurs.
La dissection des mécanismes physiopathologiques a permis d'ouvrir des pistes thérapeutiques immunomodulatrices nouvelles contre le développement des plaques d'athérosclérose et leur rupture.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
