


Rythmologie et rythmo interventionnelle
Publié le 01 jan 2020Lecture 6 min
QT long/QT court : ce qu’il faut retenir
Philippe MAURY, Anne ROLLIN, pôle cardiovasculaire et métabolique ; CHU de Toulouse - Hôpital Rangueil ; Université Toulouse III Paul Sabatier
L’intervalle QT représente l’ensemble de la dépolarisation et repolarisation de la masse myocardique ventriculaire. Il se mesure sur l’ECG depuis le début du QRS jusqu’à la fin de l’onde T, le plus souvent en D2, V2 ou V5 et plus généralement là où il est le plus long. Sa mesure cependant est délicate, car la fin de l’onde T est parfois difficile à déterminer, surtout si l’onde T est de morphologie anormale ou plate.
La fin de l’onde T est définie par l’intersection entre la ligne isoélectrique et la tangente à la pente maximale de la portion terminale descendante de l’onde T. La durée de la repolarisation dépendant étroitement de la fréquence, l’intervalle QT doit être corrigé par cette fréquence pour pouvoir être utilisé comme une variable quantitative. La formule de correction la plus utilisée reste celle de Bazett, ou le QT corrigé est égal au QT mesuré divisé par la racine carrée du cycle (intervalle entre deux QRS en secondes).
Sa valeur normale est < 440 ms chez l’homme et < 460 ms chez la femme.
Le QT long En l’absence de cause secondaire, le syndrome du QT long est défini par un score (cf. recommandations), soit par des QT corrigés > 500-480 ms ou > 480-460 ms associés à des syncopes, soit par l’existence d’une mutation pathogène (quel que soit le QT). Les causes secondaires classiques sont :
– les bradycardies ;
– les troubles ioniques (hypokaliémie, hypocalcémie, hypomagnésémie) ;
– les médications allongeant le QT, mais d’autres situations peuvent allonger le QT (hémorragie méningée, hypothyroïdie, Takotsubo par exemple).
La prévalence du QT long a été évaluée à 1 pour 2 000 sur des ECG de nouveau-nés, mais le nombre de patients atteints est probablement plus important, car le QT de base est normal dans environ 30 % des cas chez les patients mutés. Il n’y a pas de sex ratio d’un point de vue génétique puisque les mutations sont autosomiques, mais il y a plus de femmes que d’hommes avec QT long du fait d’un QT physiologiquement plus long chez la femme. Les premiers accidents rythmiques surviennent souvent dans l’enfance (mais sont possibles plus tard), plus souvent chez les garçons avant l’adolescence et plus souvent chez les femmes après.
Le diagnostic dans les formes frustes est facilité par la mesure du QT lors de manœuvres facilitatrices comme l’ECG en position debout, en phase de stress mental, à la 4-5e minute de récupération d’épreuve d’effort ou sous perfusion d’adrénaline, quoique ceci puisse être parfois peu spécifique.
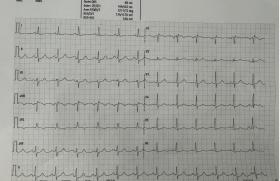
Du point de vue génétique, de nombreuses mutations ont été découvertes dans une quinzaine de gènes à ce jour, codant pour les canaux ioniques membranaires responsables de la repolarisation. Les formes les plus communes sont le LQT1 (mutation dans KCNQ1 codant pour le canal potassique IKs), LQT2 (mutations dans HeRG, canal potassique IKr) ou LQT3 (SCN5A, canal sodique INa) représentant plus de 90 % des cas. Les mutations sur les canaux potassiques sont des pertes de fonction (gêne à la repolarisation) à l’inverse des mutations sur le canal sodique (poursuite de la dépolarisation). Diverses caractéristiques ECG, cliniques, pronostiques et thérapeutiques différencient ces formes. Le LQT1 (la moitié des cas mutés) comporte des ondes T amples avec base d’implantation large (figure 1) et des accidents rythmiques essentiellement adrénergiques (effort ou émotions). Les ondes T sont plates ou crochetées dans le LQT2 (40 % des cas), avec des accidents favorisés par les à-coups adrénergiques, mais aussi les bruits intenses, parfois au repos. Les accidents sont plutôt favorisés par le repos dans le LQT3 (10 % des cas) avec une onde T acuminée à distance du QRS et un pronostic peut-être moins bon. Les autres formes sont plus exceptionnelles, mais il faut citer tout de même le syndrome d’Andersen-Tawil ou LQT7 (KCNJ2, canal IK1) avec un QT souvent peu allongé, une onde U, des TV bidirectionnelles et un meilleur pronostic, associé de manière inconstante à des anomalies morphologiques du visage et des doigts et neurologiques (paralysie périodique hypokaliémique).
Figure 1. Exemple de QT long (LQT1).
Toutes ces formes sont dans l’immense majorité des cas de transmission dominante et sans surdité (ce qu’on appelait le syndrome de Romano Ward).
Seules quelques mutations récessives dans le QT long de type 1 ou 5 peuvent donner, en cas d’atteinte homozygote, des formes très rares et graves (syndrome de Jervell-Lange Nielsen, associé à une surdité de perception bilatérale totale congénitale).
Un allongement du QT long fait courir le risque d’arythmies ventriculaires potentiellement létales appelées torsades de pointes, responsables de syncopes et pouvant dégénérer en fibrillation ventriculaire.
La mort subite est souvent précédée de syncopes ou de convulsions inexpliquées notamment lors du sport ou d’émotions, mais elle peut être inaugurale. Le risque de décès en cas de syndrome du QT long avait été évalué à 5 % par an en l’absence de traitement et est dix fois moindre depuis qu’on traite les patients par bêtabloqueur.
Avant traitement, il faut respecter les précautions d’usage (liste des médications contre-indiquées disponible sur le site Cardiogen, et lutte contre l’hypokaliémie).
• La base du traitement du QT long reste les bêtabloqueurs qui doivent être discutés dans chaque cas.
• Les bêtabloqueurs sont essentiels dans le QT long de type 1 avec une efficacité optimale quand ils sont régulièrement pris.
• Dans le QT long de type 2, leur efficacité est plus imparfaite, mais ils doivent être prescrits, la mauvaise observance étant source d’accidents.
• Les bêtabloqueurs sont plus discutés dans le QT long de type 3 et ne sont plus systématiquement prescrits à l’heure actuelle. Ils semblent efficaces dans le syndrome d’Andersen même s’ils ne font pas disparaître les salves de TV.
• Enfin ils ont démontré une certaine efficacité chez les « porteurs silencieux » (patients mutés, mais sans QT long). Le nadolol et le propanolol sont les bêtabloqueurs les plus efficaces dans le QT long, même si d’autres molécules comme le bisoprolol peuvent être proposées, à l’exception des produits à activité bêtamimétique intrinsèque, ou l’aténolol et le métoprolol qui semblent moins efficaces, et devoir être évités. Les posologies maximales tolérées doivent être prescrites. Enfin, la mexilétine (antiarythmique de classe 1) peut être proposée dans certains cas de QT long de type 3.
En cas de récidive d’arythmies malignes ou de syncopes sous bêtabloqueurs, d’autres thérapeutiques plus exceptionnelles doivent être proposées, comme la sympathectomie chirurgicale ou le défibrillateur implantable.
Le défibrillateur est en général indiqué en cas de mort subite inaugurale, et discuté dans le QT long de type 1 chez un patient jusqu’ici non traité par bêtabloqueur.
La pratique sportive notamment de compétition, jusqu’ici proscrite, semble pouvoir être autorisée dans certains cas, en accord avec le centre de compétence.
Le QT court
Il est défini par un QT corrigé < 340 ms, ou < 360 ms avec des éléments cliniques (arythmies ventriculaires malignes, mort subite et/ou QT court familial, ou mutation pathogène).
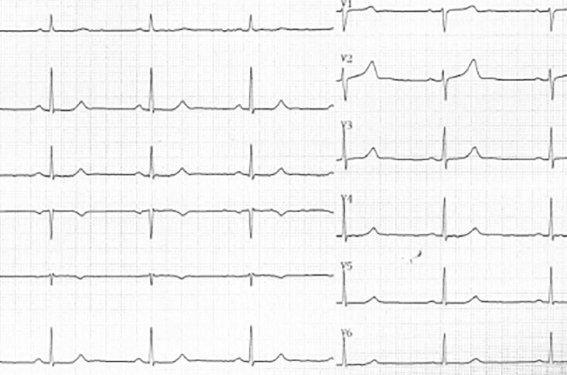
L’onde T est souvent acuminée et sans segment ST (figure 2).
Figure 2. Exemple de QT court.
Les périodes réfractaires ventriculaires et atriales sont courtes, ce qui peut justifier leur mesure dans les cas limites.
C’est un syndrome beaucoup plus rare (registres mondiaux de quelques dizaines de cas seulement) qui expose aux mêmes conséquences que le QT long. Ces patients font aussi de la fibrillation atriale à un âge jeune et ont parfois des signes de dysfonction sinusale.
Des mutations dans les mêmes gènes que le QT long ont été retrouvées (canaux potassiques, mais cette fois avec gain de fonction), mais le plus souvent actuellement l’enquête génétique reste négative.
Aucun traitement n’a fait preuve de son efficacité pour corriger ces valeurs de QT trop court, sauf la quinidine qui peut être propo sée chez les patients asymptomatiques.
Il n’y a pas de place pour la stratification du risque ou le défibrillateur en prévention primaire.
À noter que ces pathologies génétiques héréditaires doivent actuellement être prises en charge en collaboration avec les centres de compétence/référence dont la liste est disponible sur le site Cardiogen (www.filiere-cardiogen.fr).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
