




Rythmologie et rythmo interventionnelle
Publié le 22 sep 2015Lecture 13 min
Les troubles du rythme en pédiatrie
I. DENJOY, Service de cardiologie pédiatrique, hôpital Robert Debré, Paris ; Clinique Ambroise Paré, CERIC, Neuilly-sur-Seine
Les troubles du rythme de l’enfant sans malformation cardiaque associée sont rares et mettent rarement en jeu le pronostic, en dehors de certaines formes très particulières d’arythmie ventriculaire. La nature des troubles du rythme varie en fonction de l’âge.
Le diagnostic se fait essentiellement en enregistrant le trouble du rythme par l’électrocardiogramme ou des méthodes d’enregistrement continu de type Holter.
Les tachycardies régulières à QRS fins
C’est le trouble du rythme le plus fréquemment rencontré en pédiatrie. Il s’agit de tachycardies supraventriculaires. Les QRS sont fins, la fréquence cardiaque en cours de tachycardie peut dépasser 220/min chez le nourrisson. La présentation clinique dépend de l’âge et de la tolérance hémodynamique de la tachycardie.
Durant les premiers mois de vie, le trouble du rythme est parfois découvert à l’occasion d’une auscultation lors d’un examen systématique. Parfois, le diagnostic peut être fait dans un contexte d’insuffisance cardiaque aiguë, en rapport avec une cardiomyopathie rythmique méconnue. L’enregistrement électrocardiographique de bonne qualité, si possible en 50 mm/s, sur 12 dérivations, permet de suspecter le diagnostic devant une tachycardie à QRS fins avec une onde P’ rétrograde (figure 1). Le recours aux manœuvres vagales permet d’arrêter la tachycardie et de démasquer parfois, lors de la réduction, un aspect de préexcitation auriculoventriculaire. La réduction peut s’effectuer avec des manœuvres vagales (application de vessie de glace sur le visage pendant 10 secondes, ou compression des globes oculaires), ou par méthode médicamenteuse avec injection d’adénosine en flash.
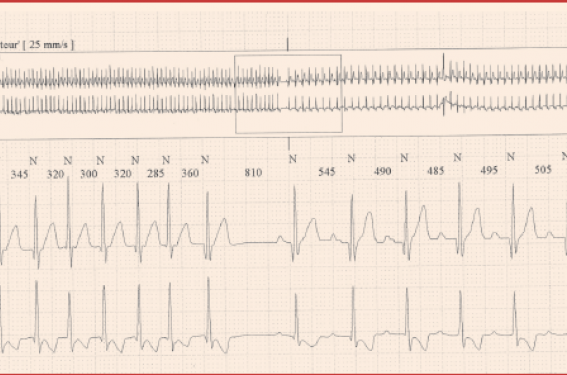
Figure 1. Réduction spontanée d’une TSV chez un nourrisson de 3 mois pendant un enregistrement Holter.
Les tachycardies jonctionnelles sur une voie accessoire auriculoventriculaire
C’est la forme la plus fréquente des tachycardies jonctionnelles, surtout lorsque les accès se produisent durant les premières années de vie (85 % des cas avant l’âge de 1 an). Il s’agit le plus souvent d’accès de tachycardie orthodromique. L’évolution des formes de présentation précoce est souvent favorable, avec une disparition des crises avant l’âge de 2 ans dans la majorité des cas. Les accès peuvent réapparaître après l’âge de 7 ans dans 30 à 40 % des cas. Ils peuvent s’accompagner chez le nourrisson d’une cardiomyopathie dilatée hypokinétique d’origine rythmique qui régresse avec la prise en charge du traitement.
Les traitements utilisés afin de prévenir les récidives sont soit des bêtabloquants, de la digoxine ou du flécaïnide, et parfois de l’amiodarone en cas de récidive avec l’un des trois traitements précédents. Le traitement est en général prescrit pour les formes à révélation précoce pendant 1 an, puis arrêté en informant les parents qu’il y a un risque éventuel de récidive, qu’il conviendra de signaler au plus vite. En cas d’accès de tachycardie récidivante chez l’enfant plus âgé, au-delà de 10 ans, des explorations électrophysiologiques doivent se discuter avec éventuellement une indication d’ablation.
En cas de persistance d’une préexcitation auriculoventriculaire patente, per manente à l’épreuve d’effort ou sur les enregistrements Holter, on peut proposer à partir de l’âge de 10 ans, surtout si l’enfant pratique du sport de façon importante, une exploration électrophysiologique qui sera éventuellement complétée, en fonction des propriétés de la voie accessoire, par une ablation.
Les tachycardies jonctionnelles par réentrée intranodale
Elles sont extrêmement rares durant les premières années de vie et apparaissent plus fréquemment après l’âge de 5-6 ans. Elles se présentent sous forme de tachycardies à QRS fins avec un intervalle RP’ extrêmement court et peuvent être traitées par des bêtabloquants si les accès sont fréquents.
Les tachycardies jonctionnelles réciproques permanentes
Les tachycardies jonctionnelles réciproques permanentes dites « tachycardie de Coumel » sont rares. Il s’agit d’une réentrée entre le noeud auriculoventriculaire et une voie accessoire à conduction lente. Leur aspect électrocardiographique est assez typique : un intervalle RP’ long avec une onde P négative en D2, D3 et aVF. Ces tachycardies, lorsqu’elles surviennent durant la première année de vie, peuvent entraîner des cardiomyopathies rythmiques (figure 2). Elles sont souvent permanentes et répondent mal aux traitements antiarythmiques. Il n’est pas inhabituel de devoir recourir à des associations comportant de l’amiodarone avec des bêtabloquants et éventuellement un antiarythmique de classe I. L’ablation par radiofréquence donne de très bons résultats dans ce cadre-là, à partir de 6 ans.
Figure 2. Rythme réciproque chronique chez un enfant de 21 mois. L’onde P’ est négative en D2-D3-aVF et V2-V3.
Les tachycardies auriculaires
Le flutter atrial
Le flutter atrial est un trouble du rythme extrêmement rare chez l’enfant en dehors de la période néonatale où il peut représenter jusqu’à 15 % des TSV. Dans la majorité des cas, le flutter survient en période néonatale, voire même en période anténatale, en l’absence de toute cardiopathie. Le trouble du rythme peut être extrêmement rapide, avec une activité atriale à 500/min et une conduction auriculoventriculaire variable, le plus souvent en 2/1 (figure 3). Le risque est la tolérance hémodynamique et il convient de réduire ces flutters le plus rapidement possible. Il est parfois nécessaire d’avoir recours à une cardioversion électrique. Une fois le trouble du rythme réduit, les risques de récidive sont très faibles et un traitement par digoxine peut être proposé pendant quelques mois puis interrompu.
Figure 3. Tachycardie à QRS fins à 200/min chez un nouveau-né. La compression oculaire (ROC) démasque une tachycardie atriale de type flutter à 500/min.
Les tachycardies atriales
Les tachycardies atriales ectopiques par automatisme anormal sont très rares durant la première année de vie. Elles sont moins rapides que les flutters auriculaires et leur diagnostic est plus difficile, pouvant entraîner en cas de non-diagnostic, une cardiomyopathie rythmique. Le pronostic est plus défavorable en cas de diagnostic précoce durant la première année de vie, alors qu’il est meilleur après l’âge de 3 ans, avec très souvent une guérison spontanée à l’adolescence. En l’absence de guérison, la réponse au traitement antiarythmique est en général médiocre, même en ayant recours à des associations comportant de l’amiodarone et des antiarythmiques de classe I. Dans ce cas, l’indication à une ablation endocavitaire peut être retenue.
L’ablation en pédiatrie
Les recommandations concernant l’ablation en pédiatrie ont fait l’objet d’un consensus européen.
Pour les enfants de plus de 5 ans, le traitement antiarythmique et l’ablation endocavitaire sont possibles en cas de tachycardie jonctionnelle compliquant un syndrome de Wolff-Parkinson-White avec accès de tachycardie.
Pour les enfants de moins de 5 ans avec tachycardie supraventriculaire non contrôlée par les antiarythmiques ou avec des effets secondaires importants liés au traitement médical, l’ablation peut être une option thérapeutique.
Chez les enfants de moins de 5 ans avec un syndrome de Wolff-Parkinson- White compliqué de tachycardie, chez les enfants entre 5 et 10 ans, avec une préexcitation auriculoventriculaire asymptomatique et en cas de tachycardie supraventriculaire peu fréquente, après 5 ans, l’ablation est une possibilité à discuter avec les parents et les équipes.
Les troubles du rythme ventriculaire
Ils sont très rares chez l’enfant en dehors de toute cardiopathie. Il existe des tachycardies ventriculaires monomorphes souvent de bon pronostic, et des tachycardies ventriculaires polymorphes, avec pronostic vital engagé, compliquant souvent une canalopathie.
Les tachycardies ventriculaires monomorphes
Elles sont rares chez l’enfant. La forme la plus fréquente est la tachycardie ventriculaire en salves avec un aspect de retard gauche, axe inférieur (figure 4). Dans cette forme, il existe des extrasystoles ventriculaires isolées, avec des doublets et des salves plus ou moins soutenues. La fréquence lors des salves n’est pas très rapide, en moyenne à 150-160/min. L’échographie permet d’éliminer une cardiopathie associée. Les extrasystoles isolées disparaissent souvent à l’effort. Elles répondent en général bien aux bêtabloquants qui seront indiqués si l’enfant se plaint de palpitations.
Figure 4. Tachycardie ventriculaire de type retard droit sur un ECG 12 dérivations, sensible au vérapamil, chez une enfant de 10 ans.
Il existe une autre forme de tachycardie ventriculaire dite « tachycardie sensible au vérapamil » dont la morphologie est caractérisée par un aspect de retard droit, axe ascendant. Les QRS sont en général assez fins et le diagnostic est parfois fait à l’occasion d’une manœuvre vagale permettant d’identifier la dissociation auriculoventriculaire. Cette tachycardie répond particulièrement bien aux inhibiteurs calciques qui permettent sa réduction par voie intraveineuse. Un traitement préventif au long cours est assuré par un traitement inhibiteur calcique ou par une ablation qui donne de très bons résultats.
Les tachycardies ventriculaires polymorphes de type torsades de pointes ou fibrillation ventriculaire
En l’absence de toute cardiopathie, ces tachycardies ventriculaires polymorphes sont la forme la plus grave de troubles du rythme de l’enfant et l’exposent au risque de mort subite. Il convient après leur réduction par choc électrique de faire un bilan étiologique. Les principales causes de ces tachycardies sont le syndrome du QT long congénital, les tachycardies ventriculaires polymorphes et le syndrome de Brugada.
Le syndrome du QT long congénital
Le syndrome du QT long congénital se caractérise par un allongement de la repolarisation ventriculaire (intervalle QTc), associé à un risque de trouble du rythme ventriculaire grave (torsades de pointe et fibrillation ventriculaire) entraînant des syncopes et éventuellement une mort subite.
L’intervalle QT est considéré comme allongé si le QTc est supérieur à 440 ms selon la formule de Bazett (QTc = QT/√RR). La prévalence du syndrome du QT long congénital est estimée à 1/2 000 et c’est l’une des principales causes de mort subite chez le sujet jeune. Il s’agit d’une canalopathie en rapport avec une mutation dans l’un des gènes intervenant dans l’électrophysiologie cellulaire. À l’heure actuelle, 16 gènes codant principalement pour des canaux ioniques cardiaques ont été identifiés. Les études de biologie moléculaire montrent que dans 80 % des formes génétiques identifiées, trois principaux gènes sont en cause : KCNQ1 (environ 50 % des formes) correspondant à la forme QT long type 1, KCNH2 (QT long de type 2 ; 40 %) et SCN5A (10 % ; forme QT long type 3).
Quelle que soit la forme génétique, le syndrome du QT long congénital a des caractéristiques communes : allongement de l’intervalle QTc, risque de syncope et de mort subite par trouble du rythme ventriculaire polymorphe de type torsades de pointes. Il existe des caractéristiques spécifiques pour chacune des principales formes génétiques de QT long congénital concernant l’aspect électrocardiographique (figure 5), le risque de complication rythmique et les événements favorisants ces troubles du rythme graves. Les événements cardiaques surviennent principalement dans l’enfance et les séries rapportent la survenue de syncope ou de mort subite dans plus de 60 % des cas avant l’âge de 8 ans.
Figure 5. Morphologie de l’onde T selon les formes génétiques LQT1, LQT2 et LQT3.
Le diagnostic de QT long congénital
Il repose sur l’enregistrement du trouble du rythme ventriculaire (mais rare), l’allongement de l’intervalle QTc, l’anomalie de morphologie de l’onde T et l’identification d’une anomalie génétique lorsque cela est possible. Les critères diagnostiques du syndrome du QT long congénital ont été précisés dans un consensus récent en 2013, avec un score diagnostique établi à partir des principales caractéristiques électrocardiographiques et cliniques du syndrome.
Le syndrome du QT long congénital de type 1 peut être suspecté chez le nourrisson, voire chez le fœtus, en cas de bradycardie sinusale inappropriée en l’absence de toute anomalie de conduction. Les syncopes et accidents rythmiques surviennent essentiellement dans un contexte d’effort, tout particulièrement en cas de sport nautique.
Le syndrome du QT long congénital de type 2 se complique de syncope et de troubles du rythme ventriculaire à l’occasion d’émotions fortes (réveil nocturne, par exemple). L’onde T sur l’électrocardiogramme a un aspect en double bosse assez typique. La présentation néonatale de ce syndrome associe volontiers un trouble de conduction auriculoventriculaire fonctionnel de type BAV 2/1.
Le syndrome du QT long congénital de type 3, est le plus rare. Les syncopes et accidents rythmiques surviennent essentiellement en période de sommeil ou au repos. Sur l’électrocardiogramme, l’onde T est très ample, et d’apparition tardive. Les accidents rythmiques sont moins fréquents qu’avec les formes QT1 et QT2, mais plus souvent mortels.
Le traitement du syndrome du QT long congénital
Quelle que soit la forme génétique, il repose sur la mise en place d’un traitement préventif strict par bêtabloquants, généralement le nadolol à la posologie de 50 mg/m2 en deux prises.
Chez le nourrisson, on aura recours au propranolol à la posologie moyenne de 2 à 5 mg/kg en 3 prises. Une liste de médicaments susceptibles d’aggraver les anomalies de la repolarisation ventriculaire doit être fournie aux parents et aux pédiatres. Certaines modifications dans le mode de vie doivent également être recommandées (interdiction du sport de compétition, de la natation, changement des sonneries de réveil matin). Une grande majorité des enfants avec un syndrome du QT long congénital répondent bien aux bêtabloquants et à la prise en compte des situations cliniques favorisant la survenue des troubles du rythme. Les indications de défibrillateur cardiaque restent rares, elles concernent principalement certaines formes de QT 3 ou de QT 2.
Les tachycardies ventriculaires catécholergiques
Les tachycardies ventriculaires catécholergiques sont une cause rare de trouble du rythme ventriculaire malin chez l’enfant de plus de 3 ans. Le diagnostic est difficile car l’électrocardiogramme, en dehors des moments symptomatiques, est normal. Le diagnostic doit être suspecté devant une syncope dans un contexte d’effort ou d’émotions pouvant s’accompagner éventuellement de convulsions.
Le diagnostic a fait l’objet d’un consensus récent et repose sur la mise en évidence de troubles du rythme ventriculaires polymorphes à l’effort enregistrés à l’occasion d’un Holter, d’une épreuve d’effort (figure 6) ou d’un test à l’isoprénaline. Au-delà d’une fréquence sinusale supérieure à 130/min apparaissent des extrasystoles ventriculaires polymorphes, d’abord isolées, puis bigéminées, puis en salves, plus ou moins soutenues et plus ou moins rapides. À l’arrêt de l’effort ou de la perfusion d’isoprénaline, la fréquence sinusale se ralentit et les troubles du rythme disparaissent dans l’ordre inverse où ils sont apparus. Il s’agit d’une canalopathie mettant en jeu la régulation du calcium intracellulaire. Les principaux gènes impliqués sont les gènes RyR2, CASQ2 et plus récemment la Triadine. Une anomalie génétique est retrouvée dans 70 % des cas lorsque le bilan est effectué. Le traitement consiste à interdire tous sports de compétition, tous sports aquatiques, et à limiter l’activité physique de telle façon qu’en association avec le traitement bêtabloquant, les fréquences sinusales ne dépassent pas 130/min. Les doses de bêtabloquant nécessaires pour contrôler l’arythmie sont souvent élevées, 75 voire 100 mg/m2 en deux prises de nadolol. Les arythmies ventriculaires peuvent persister malgré le traitement bêtabloquant à forte dose et nécessitent dans ce cas l’adjonction d’un traitement par flécaïnide. En cas de récidive de syncope, malgré un traitement antiarythmique bien conduit, il faudra discuter la mise en place d’un défibrillateur automatique implantable.
Figure 6. Épreuve d’effort chez un enfant de 10 ans, réalisée pour syncope, montrant des ESV polymorphes avec doublets et salves brèves.
Le syndrome de Brugada
Le syndrome de Brugada a été décrit dans les années 1990. Il se caractérise par une anomalie de la repolarisation et des précordiales droites associé à un risque de mort subite par fibrillation ventriculaire survenant dans un contexte de repos ou au sommeil. Il existe une dizaine de gènes impliqués dans ce syndrome, le gène SCN5A étant le plus fréquemment impliqué (dans 20 à 25 % des cas). Le risque de mort subite est plus important aux alentours de 40 ans, chez les hommes. Le diagnostic est posé en cas de sus-décalage du segment ST convexe dans les dérivations V1 et V2, enregistré soit de façon spontanée, soit à l’occasion d’un test pharmacologique (ajmaline, flécaïnide). Cet aspect est fluctuant et peut être également démasqué à l’occasion d’épisodes fébriles.
Les formes pédiatriques sont extrême ment rares et peuvent être découvertes à l’occasion de bilans familiaux ou à l’occasion d’une syncope, dans un contexte fébrile. Les formes symptomatiques avec anomalies électrocardiographiques de type 1 et syncope, diagnostiquées chez les enfants de moins de 15 ans, sont rares mais graves. À l’opposé, le syndrome de Brugada diagnostiqué à l’occasion des bilans familiaux qui sont proposés chez les enfants après l’âge de 15 ans, en l’absence de tout symptôme et avec un électrocardiogramme de surface normal, semble avoir un bon pronostic.
Une prise en charge familiale !
Dans la mesure où ces syndromes (syndrome du QT long congénital, syndrome de Brugada et tachycardie ventriculaire catécholergique), ont un substrat génétique et obéissent à des modèles de transmission autosomique dominante, il convient lorsque le diagnostic est porté chez un enfant, d’examiner ses parents et ses frères et sœurs en mettant en place une stratégie de diagnostic spécifique selon le syndrome.
Les enfants et leur famille doivent être adressés dans les centres de référence nationaux ou les centres de compétence afin de pouvoir bénéficier d’un bilan clinique cardiologique et génétique spécifique, permettant de mettre en place la prise en charge initiale et les modalités du suivi.
Conclusion
Les troubles du rythme chez l’enfant sont rares en l’absence de cardiopathie.
Il s’agit majoritairement de tachycardies supraventriculaires qui répondent bien au traitement médical et disparaissent le plus souvent avec l’âge.
Les troubles du rythme ventriculaires peuvent mettre en jeu le pronostic vital et révéler une maladie rythmique héréditaire d’origine génétique nécessitant une prise en charge spécialisée dans les centres de référence.
"Publié dans RythmologieS"
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
